Cheng Feixiong, Jia Peilin, Wang Quan, Zhao Zhongming
Department of Biomedical Informatics, Vanderbilt University School of Medicine, Nashville, Tennessee, USA.
Department of Biomedical Informatics, Vanderbilt University School of Medicine, Nashville, Tennessee, USA; Center for Quantitative Sciences, Vanderbilt University Medical Center, Nashville, Tennessee, USA; Department of Cancer Biology, Vanderbilt University School of Medicine, Nashville, Tennessee, USA; Department of Psychiatry, Vanderbilt University School of Medicine, Nashville, Tennessee, USA.
Oncotarget. 2014 Jun 15;5(11):3697-710. doi: 10.18632/oncotarget.1984.
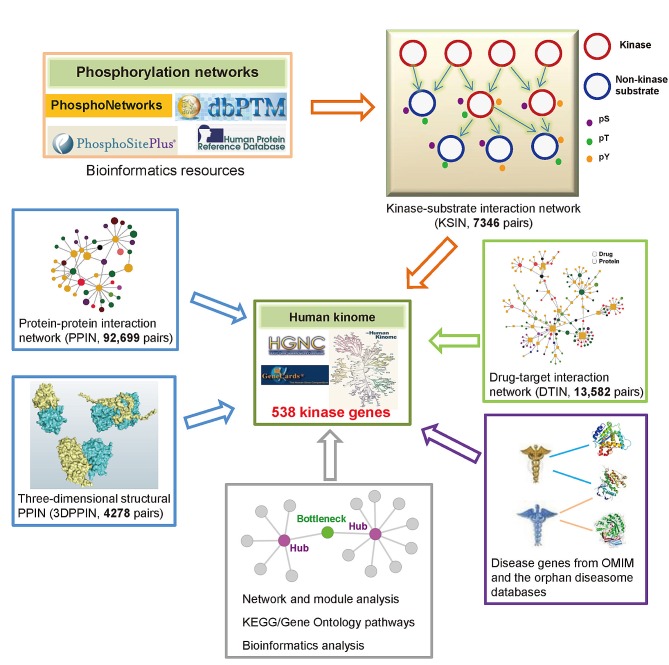
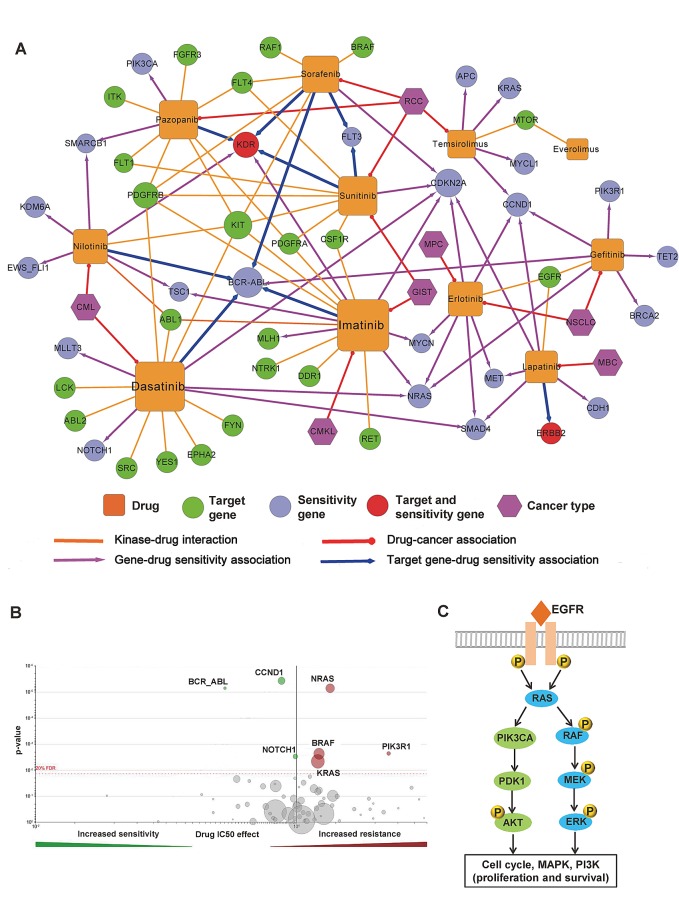
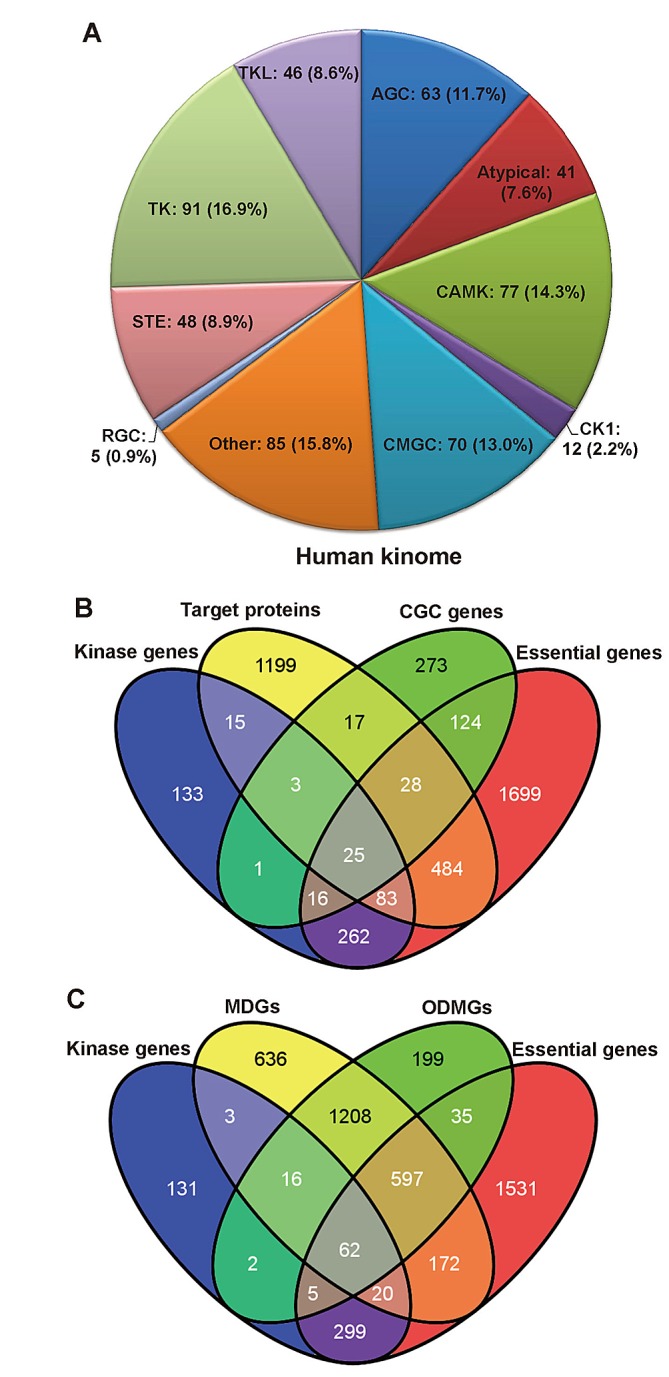
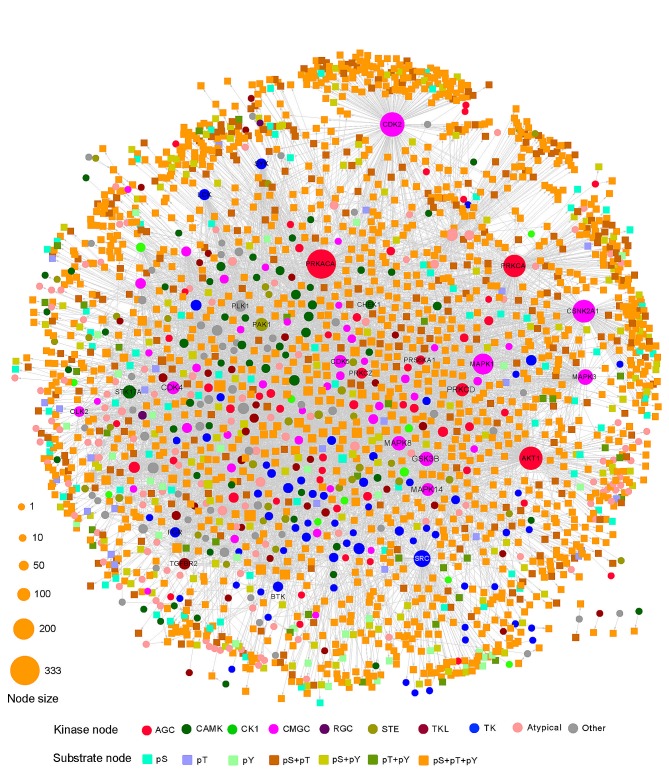
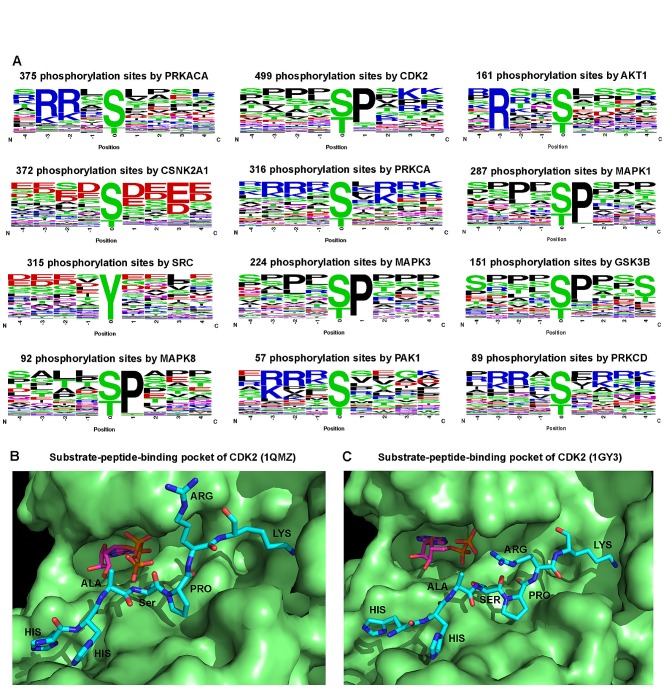
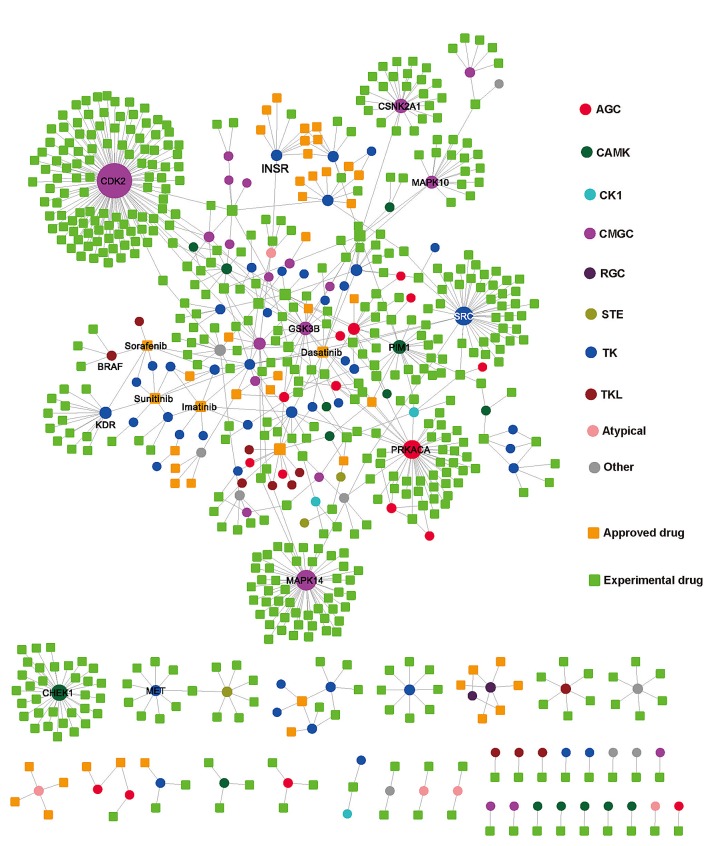
The human kinome is gaining importance through its promising cancer therapeutic targets, yet no general model to address the kinase inhibitor resistance has emerged. Here, we constructed a systems biology-based framework to catalogue the human kinome, including 538 kinase genes, in the broader context of the human interactome. Specifically, we constructed three networks: a kinase-substrate interaction network containing 7,346 pairs connecting 379 kinases to 36,576 phosphorylation sites in 1,961 substrates, a protein-protein interaction network (PPIN) containing 92,699 pairs, and an atomic resolution PPIN containing 4,278 pairs. We identified the conserved regulatory phosphorylation motifs (e.g., Ser/Thr-Pro) using a sequence logo analysis. We found the typical anticancer target selection strategy that uses network hubs as drug targets, might lead to a high adverse drug reaction risk. Furthermore, we found the distinct network centrality of kinases creates a high anticancer drug resistance risk by feedback or crosstalk mechanisms within cellular networks. This notion is supported by the systematic network and pathway analyses that anticancer drug resistance genes are significantly enriched as hubs and heavily participate in multiple signaling pathways. Collectively, this comprehensive human kinome interactome map sheds light on anticancer drug resistance mechanisms and provides an innovative resource for rational kinase inhibitor design.
人类激酶组凭借其颇具前景的癌症治疗靶点而愈发重要,但尚未出现解决激酶抑制剂耐药性问题的通用模型。在此,我们构建了一个基于系统生物学的框架,以便在人类相互作用组的更广泛背景下对包含538个激酶基因的人类激酶组进行编目。具体而言,我们构建了三个网络:一个激酶 - 底物相互作用网络,包含7346对连接379个激酶与1961个底物中36576个磷酸化位点的相互作用;一个包含92699对相互作用的蛋白质 - 蛋白质相互作用网络(PPIN),以及一个包含4278对相互作用的原子分辨率PPIN。我们使用序列标志分析确定了保守的调节性磷酸化基序(例如,丝氨酸/苏氨酸 - 脯氨酸)。我们发现,将网络枢纽用作药物靶点的典型抗癌靶点选择策略可能会导致较高的药物不良反应风险。此外,我们发现激酶独特的网络中心性通过细胞网络内的反馈或串扰机制产生了较高的抗癌药物耐药性风险。系统的网络和通路分析支持了这一观点,即抗癌药物耐药基因作为枢纽显著富集,并大量参与多种信号通路。总体而言,这张全面的人类激酶组相互作用组图谱揭示了抗癌药物耐药机制,并为合理设计激酶抑制剂提供了创新资源。