Brodie Aharon, Tovia-Brodie Oholi, Ofran Yanay
The Goodman Faculty of Life Sciences, Nanotechnology Building, Bar Ilan University, Ramat Gan, Israel.
Department of Cardiology, Tel-Aviv Sourasky Medical Center, Tel-Aviv, Israel.
PLoS One. 2014 Jul 9;9(7):e100887. doi: 10.1371/journal.pone.0100887. eCollection 2014.
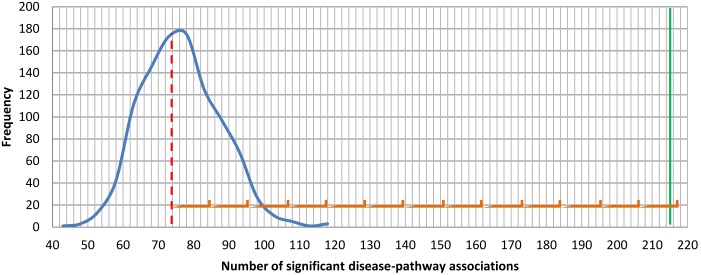
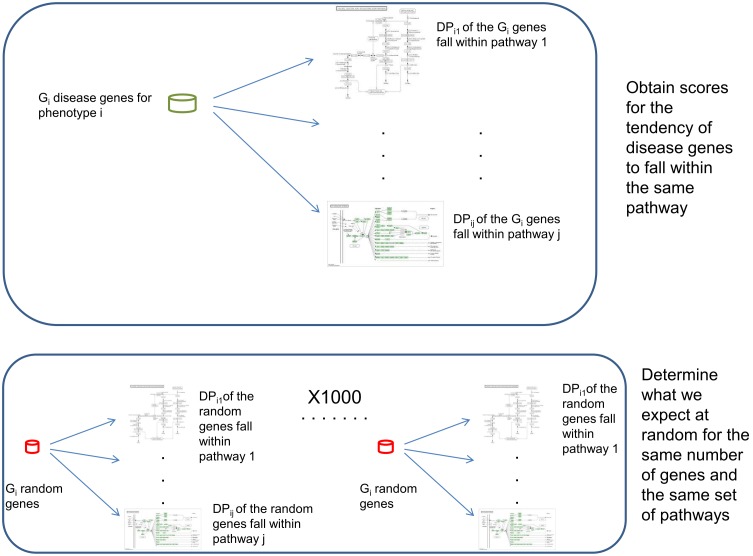
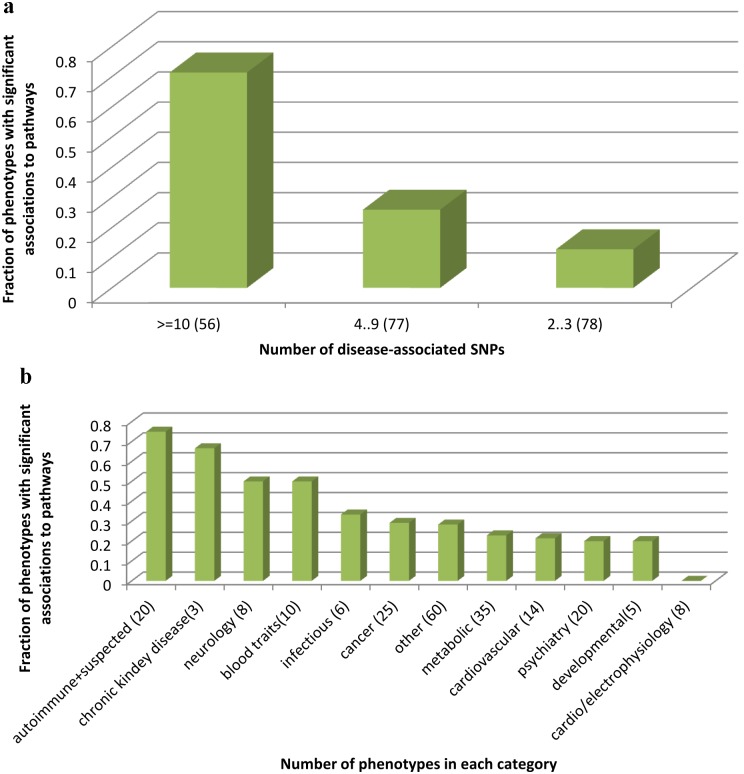
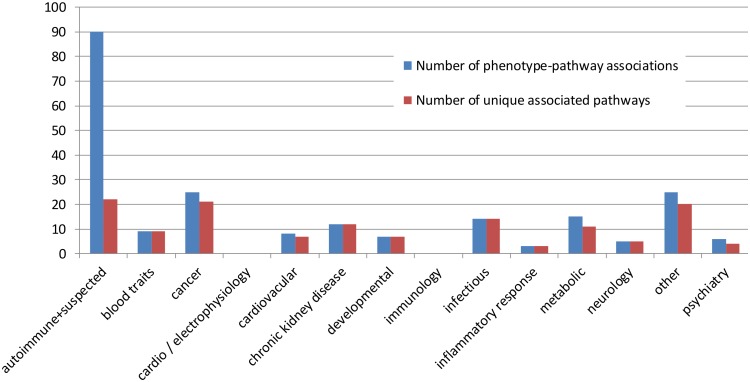
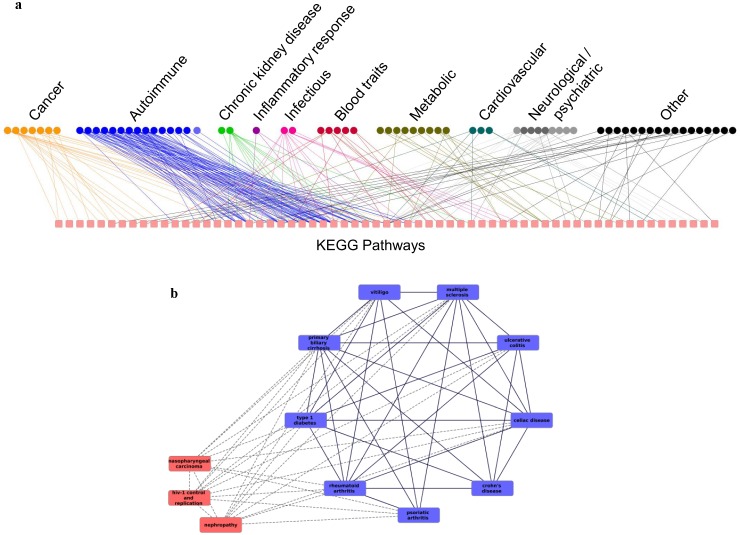
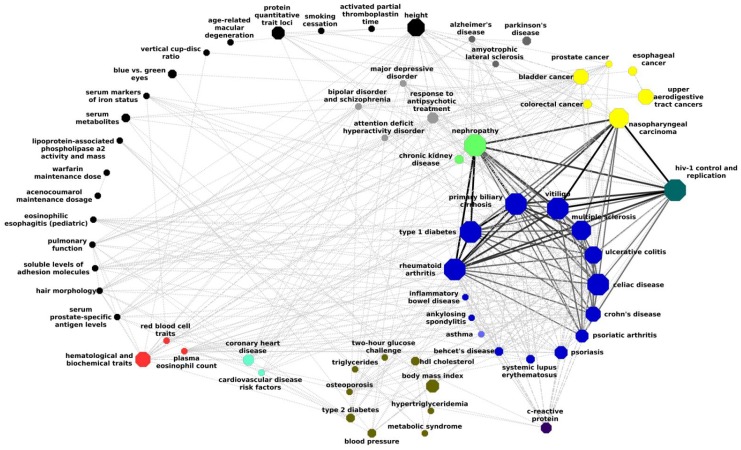
The widely used pathway-based approach for interpreting Genome Wide Association Studies (GWAS), assumes that since function is executed through the interactions of multiple genes, different perturbations of the same pathway would result in a similar phenotype. This assumption, however, was not systemically assessed on a large scale. To determine whether SNPs associated with a given complex phenotype affect the same pathways more than expected by chance, we analyzed 368 phenotypes that were studied in >5000 GWAS. We found 216 significant phenotype-pathway associations between 70 of the phenotypes we analyzed and known pathways. We also report 391 strong phenotype-phenotype associations between phenotypes that are affected by the same pathways. While some of these associations confirm previously reported connections, others are new and could shed light on the molecular basis of these diseases. Our findings confirm that phenotype-associated SNPs cluster into pathways much more than expected by chance. However, this is true for <20% (70/368) of the phenotypes. Different types of phenotypes show markedly different tendencies: Virtually all autoimmune phenotypes show strong clustering of SNPs into pathways, while most cancers and metabolic conditions, and all electrophysiological phenotypes, could not be significantly associated with any pathway despite being significantly associated with a large number of SNPs. While this may be due to missing data, it may also suggest that these phenotypes could result only from perturbations of specific genes and not from other perturbations of the same pathway. Further analysis of pathway-associated versus gene-associated phenotypes is, therefore, needed in order to understand disease etiology and in order to promote better drug target selection.
广泛使用的基于通路的方法来解释全基因组关联研究(GWAS),假定由于功能是通过多个基因的相互作用来执行的,同一通路的不同扰动会导致相似的表型。然而,这一假设尚未在大规模上进行系统评估。为了确定与给定复杂表型相关的单核苷酸多态性(SNP)是否比偶然预期的更多地影响相同的通路,我们分析了在超过5000项GWAS中研究的368种表型。我们在分析的70种表型与已知通路之间发现了216个显著的表型-通路关联。我们还报告了受相同通路影响的表型之间的391个强表型-表型关联。虽然其中一些关联证实了先前报道的联系,但其他一些是新的,可能会揭示这些疾病的分子基础。我们的研究结果证实,与表型相关的SNP聚集在通路中的程度远远超过偶然预期。然而,对于不到20%(70/368)的表型来说是这样。不同类型的表型显示出明显不同的趋势:几乎所有自身免疫性表型都显示SNP在通路中强烈聚集,而大多数癌症和代谢状况以及所有电生理表型,尽管与大量SNP显著相关,但与任何通路都没有显著关联。虽然这可能是由于数据缺失,但也可能表明这些表型可能仅由特定基因的扰动导致,而不是同一通路的其他扰动。因此,需要进一步分析与通路相关的表型和与基因相关的表型,以便了解疾病病因并促进更好的药物靶点选择。