Roosing Susanne, Lamers Ideke J C, de Vrieze Erik, van den Born L Ingeborgh, Lambertus Stanley, Arts Heleen H, Peters Theo A, Hoyng Carel B, Kremer Hannie, Hetterschijt Lisette, Letteboer Stef J F, van Wijk Erwin, Roepman Ronald, den Hollander Anneke I, Cremers Frans P M
Department of Human Genetics, Radboud University Medical Center, PO Box 9101, 6500 HB Nijmegen, the Netherlands; Radboud Institute for Molecular Life Sciences, Radboud University Nijmegen, PO Box 9101, 6500 HB Nijmegen, the Netherlands.
Department of Human Genetics, Radboud University Medical Center, PO Box 9101, 6500 HB Nijmegen, the Netherlands; Department of Otorhinolaryngology, Radboud University Medical Center, PO Box 9101, 6500 HB Nijmegen, the Netherlands.
Am J Hum Genet. 2014 Aug 7;95(2):131-42. doi: 10.1016/j.ajhg.2014.06.012. Epub 2014 Jul 10.
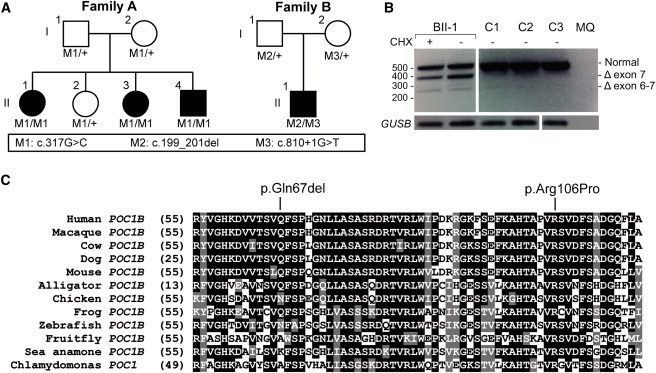
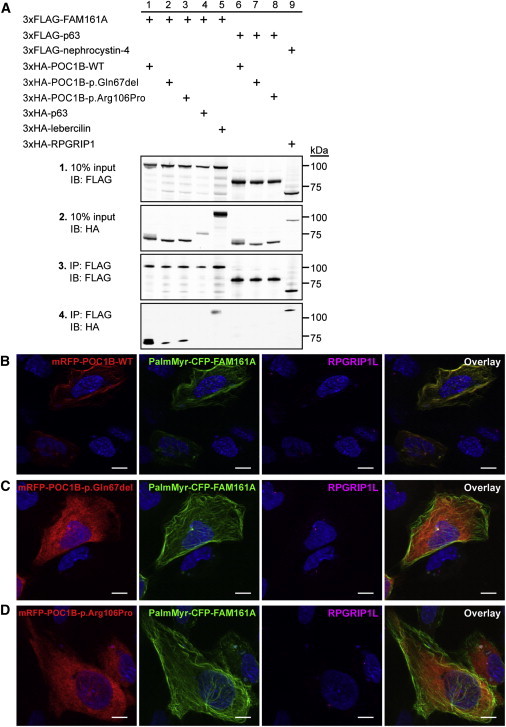
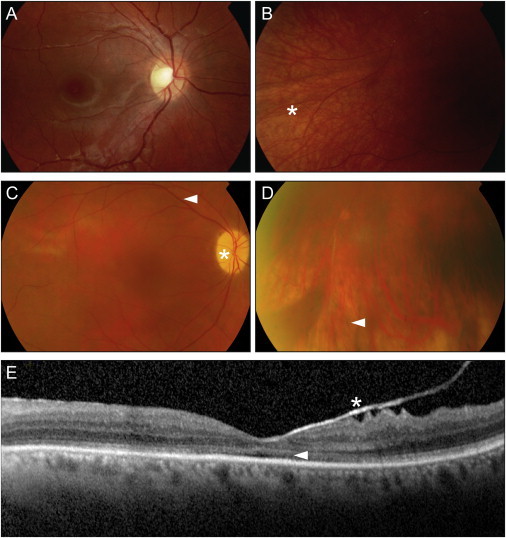
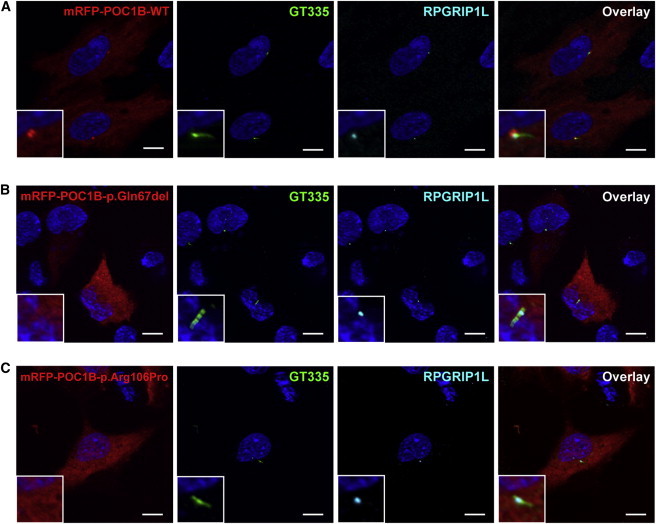
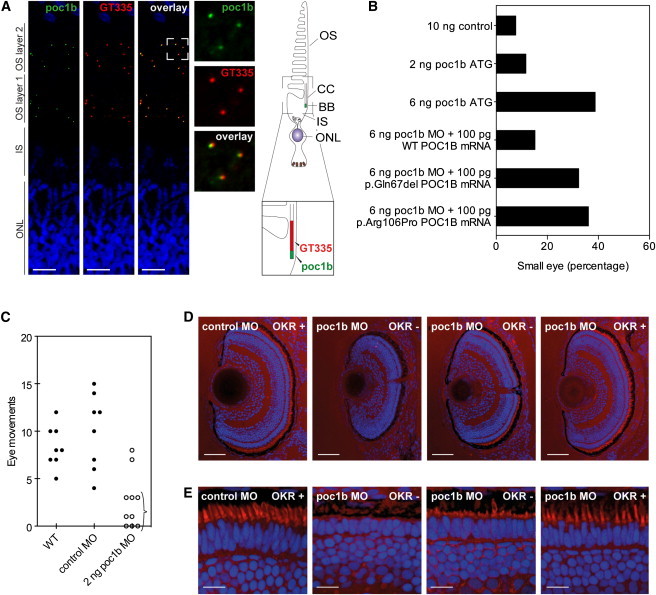
Exome sequencing revealed a homozygous missense mutation (c.317C>G [p.Arg106Pro]) in POC1B, encoding POC1 centriolar protein B, in three siblings with autosomal-recessive cone dystrophy or cone-rod dystrophy and compound-heterozygous POC1B mutations (c.199_201del [p.Gln67del] and c.810+1G>T) in an unrelated person with cone-rod dystrophy. Upon overexpression of POC1B in human TERT-immortalized retinal pigment epithelium 1 cells, the encoded wild-type protein localized to the basal body of the primary cilium, whereas this localization was lost for p.Arg106Pro and p.Gln67del variant forms of POC1B. Morpholino-oligonucleotide-induced knockdown of poc1b translation in zebrafish resulted in a dose-dependent small-eye phenotype, impaired optokinetic responses, and decreased length of photoreceptor outer segments. These ocular phenotypes could partially be rescued by wild-type human POC1B mRNA, but not by c.199_201del and c.317C>G mutant human POC1B mRNAs. Yeast two-hybrid screening of a human retinal cDNA library revealed FAM161A as a binary interaction partner of POC1B. This was confirmed in coimmunoprecipitation and colocalization assays, which both showed loss of FAM161A interaction with p.Arg106Pro and p.Gln67del variant forms of POC1B. FAM161A was previously implicated in autosomal-recessive retinitis pigmentosa and shown to be located at the base of the photoreceptor connecting cilium, where it interacts with several other ciliopathy-associated proteins. Altogether, this study demonstrates that POC1B mutations result in a defect of the photoreceptor sensory cilium and thus affect cone and rod photoreceptors.
外显子组测序在三名患有常染色体隐性遗传性视锥细胞营养不良或视锥-视杆细胞营养不良的兄弟姐妹中发现,编码POC1中心粒蛋白B的POC1B基因存在纯合错义突变(c.317C>G [p.Arg106Pro]);在一名患有视锥-视杆细胞营养不良的无关个体中发现了复合杂合性POC1B突变(c.199_201del [p.Gln67del]和c.810+1G>T)。在人端粒酶逆转录酶永生化视网膜色素上皮1细胞中过表达POC1B时,编码的野生型蛋白定位于初级纤毛的基体,而对于POC1B的p.Arg106Pro和p.Gln67del变异形式,这种定位则消失。在斑马鱼中,吗啉代寡核苷酸诱导的poc1b翻译敲低导致剂量依赖性小眼睛表型、视动反应受损以及光感受器外段长度缩短。这些眼部表型可部分由野生型人POC1B mRNA挽救,但不能由c.199_201del和c.317C>G突变型人POC1B mRNA挽救。对人视网膜cDNA文库进行酵母双杂交筛选发现,FAM161A是POC1B的二元相互作用伙伴。这在共免疫沉淀和共定位试验中得到了证实,这两项试验均显示FAM161A与POC1B的p.Arg106Pro和p.Gln67del变异形式的相互作用丧失。FAM161A先前被认为与常染色体隐性遗传性视网膜色素变性有关,并显示位于光感受器连接纤毛的基部,在那里它与其他几种与纤毛病相关的蛋白质相互作用。总之,这项研究表明,POC1B突变导致光感受器感觉纤毛缺陷,从而影响视锥和视杆光感受器。