Plongthongkum Nongluk, van Eijk Kristel R, de Jong Simone, Wang Tina, Sul Jae Hoon, Boks Marco P M, Kahn René S, Fung Ho-Lim, Ophoff Roel A, Zhang Kun
Department of Bioengineering, University of California San Diego, La Jolla, California, United States of America.
Department of Psychiatry, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, Utrecht, The Netherlands.
PLoS One. 2014 Jul 14;9(7):e99313. doi: 10.1371/journal.pone.0099313. eCollection 2014.
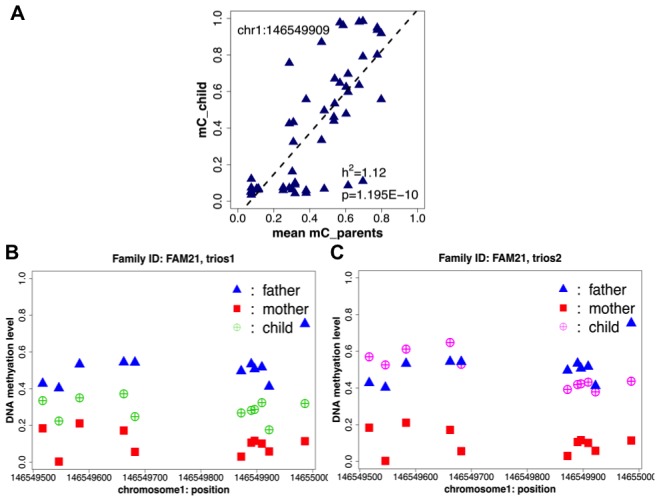
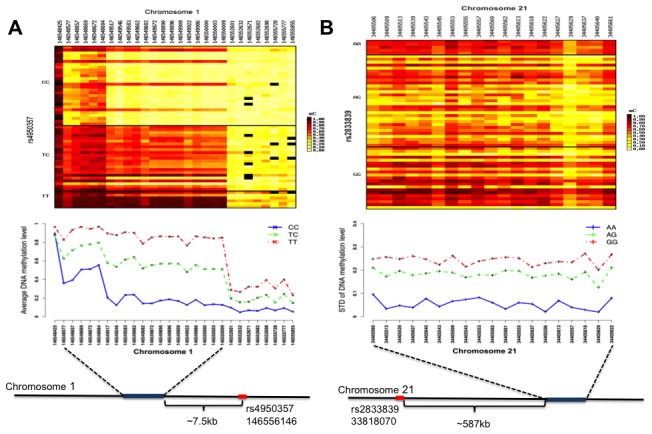
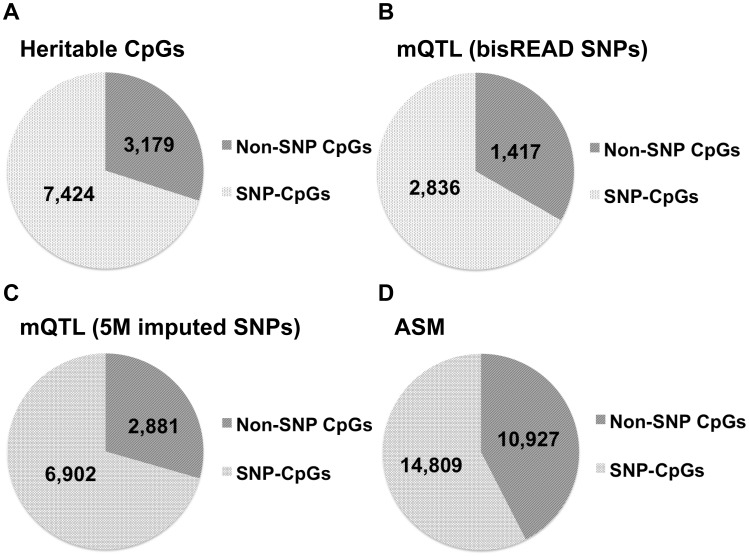
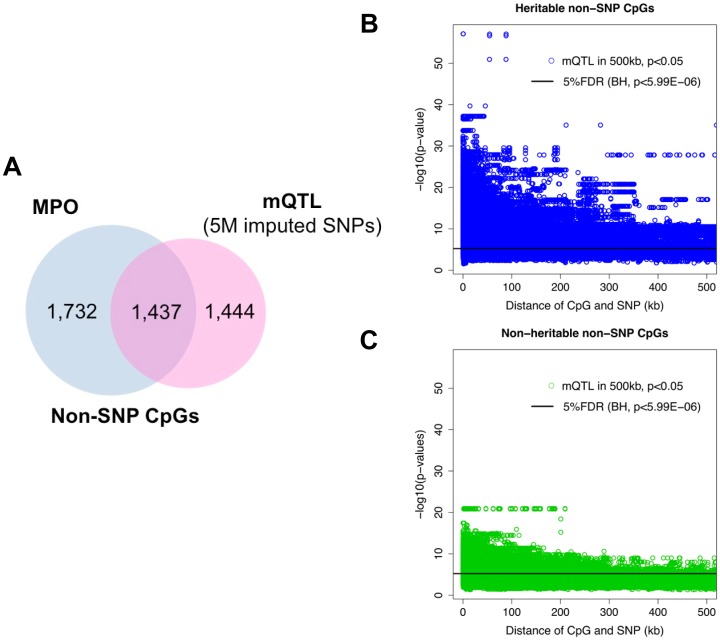
Genetic polymorphisms can shape the global landscape of DNA methylation, by either changing substrates for DNA methyltransferases or altering the DNA binding affinity of cis-regulatory proteins. The interactions between CpG methylation and genetic polymorphisms have been previously investigated by methylation quantitative trait loci (mQTL) and allele-specific methylation (ASM) analysis. However, it remains unclear whether these approaches can effectively and comprehensively identify all genetic variants that contribute to the inter-individual variation of DNA methylation levels. Here we used three independent approaches to systematically investigate the influence of genetic polymorphisms on variability in DNA methylation by characterizing the methylation state of 96 whole blood samples in 52 parent-child trios from 22 nuclear pedigrees. We performed targeted bisulfite sequencing with padlock probes to quantify the absolute DNA methylation levels at a set of 411,800 CpG sites in the human genome. With mid-parent offspring analysis (MPO), we identified 10,593 CpG sites that exhibited heritable methylation patterns, among which 70.1% were SNPs directly present in methylated CpG dinucleotides. We determined the mQTL analysis identified 49.9% of heritable CpG sites for which regulation occurred in a distal cis-regulatory manner, and that ASM analysis was only able to identify 5%. Finally, we identified hundreds of clusters in the human genome for which the degree of variation of CpG methylation, as opposed to whether or not CpG sites were methylated, was associated with genetic polymorphisms, supporting a recent hypothesis on the genetic influence of phenotypic plasticity. These results show that cis-regulatory SNPs identified by mQTL do not comprise the full extent of heritable CpG methylation, and that ASM appears overall unreliable. Overall, the extent of genome-methylome interactions is well beyond what is detectible with the commonly used mQTL and ASM approaches, and is likely to include effects on plasticity.
基因多态性可以通过改变DNA甲基转移酶的底物或改变顺式调控蛋白的DNA结合亲和力来塑造DNA甲基化的全局格局。此前,通过甲基化数量性状位点(mQTL)和等位基因特异性甲基化(ASM)分析,对CpG甲基化与基因多态性之间的相互作用进行了研究。然而,尚不清楚这些方法能否有效且全面地识别出所有导致个体间DNA甲基化水平差异的基因变异。在此,我们使用三种独立的方法,通过对来自22个核心家系的52个亲子三联体中的96个全血样本的甲基化状态进行表征,系统地研究了基因多态性对DNA甲基化变异性的影响。我们使用锁式探针进行靶向亚硫酸氢盐测序,以量化人类基因组中一组411,800个CpG位点的绝对DNA甲基化水平。通过中亲后代分析(MPO),我们鉴定出10,593个呈现可遗传甲基化模式的CpG位点,其中70.1%是直接存在于甲基化CpG二核苷酸中的单核苷酸多态性(SNP)。我们确定mQTL分析识别出49.9%的可遗传CpG位点,其调控以远端顺式调控方式发生,而ASM分析仅能识别5%。最后,我们在人类基因组中鉴定出数百个簇,其中CpG甲基化的变异程度而非CpG位点是否甲基化与基因多态性相关,这支持了最近关于表型可塑性的遗传影响的假说。这些结果表明,mQTL鉴定出的顺式调控SNP并不涵盖可遗传CpG甲基化的全部范围,并且ASM总体上似乎不可靠。总体而言,基因组 - 甲基化组相互作用的程度远远超出了常用的mQTL和ASM方法所能检测到的范围,并且可能包括对可塑性的影响。