Skewes-Cox Peter, Sharpton Thomas J, Pollard Katherine S, DeRisi Joseph L
Biological and Medical Informatics Graduate Program, University of California San Francisco, San Francisco, California, United States of America; Departments of Medicine, Biochemistry and Biophysics, and Microbiology, University of California San Francisco, San Francisco, California, United States of America; Howard Hughes Medical Institute, Bethesda, Maryland, United States of America.
The J. David Gladstone Institutes, University of California San Francisco, San Francisco, California, United States of America.
PLoS One. 2014 Aug 20;9(8):e105067. doi: 10.1371/journal.pone.0105067. eCollection 2014.
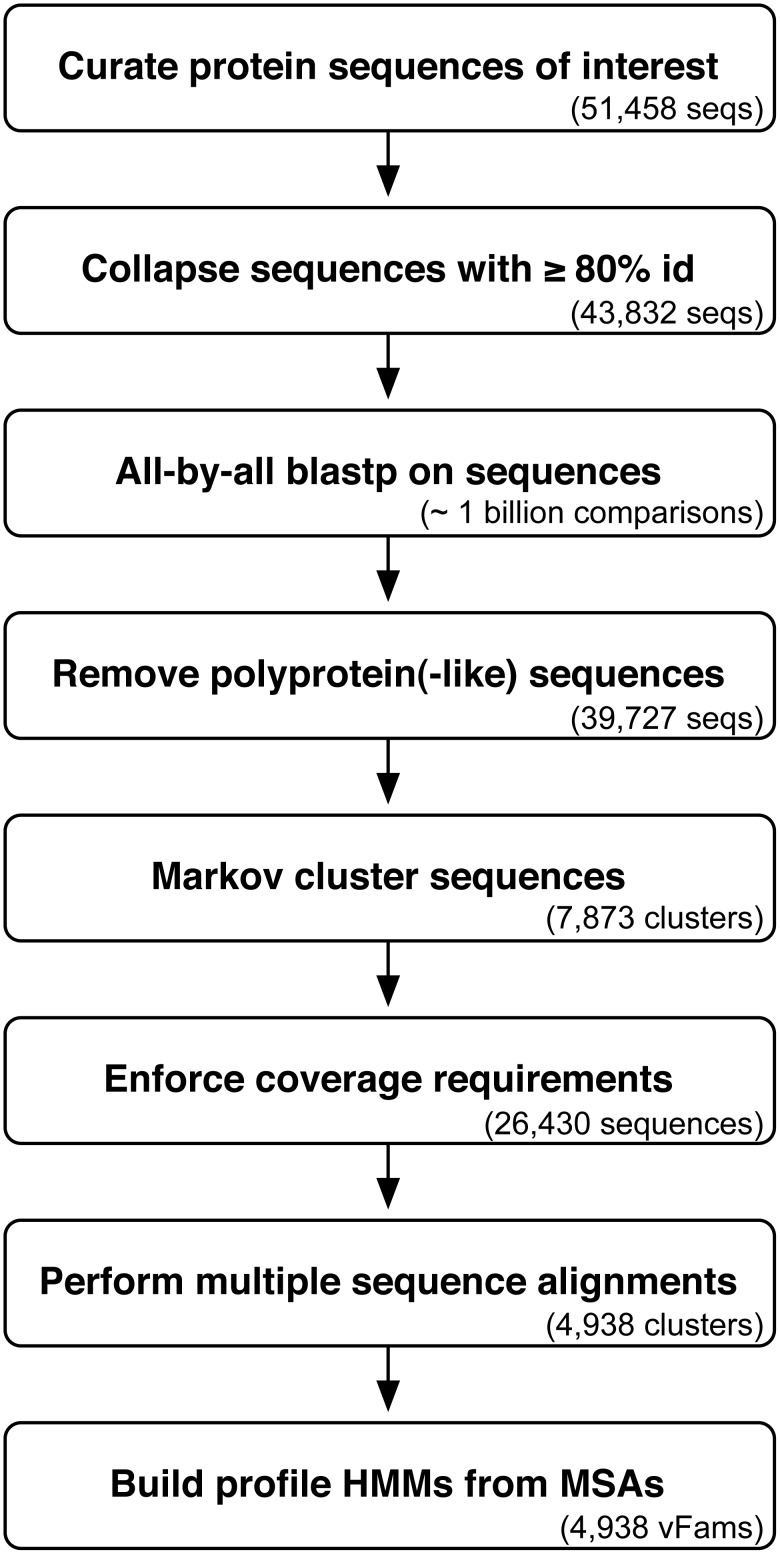
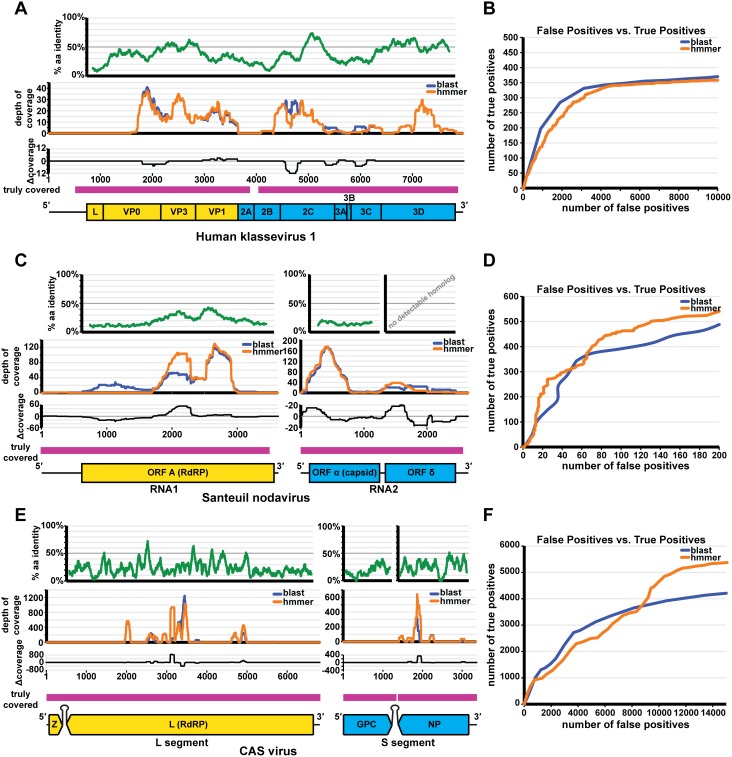
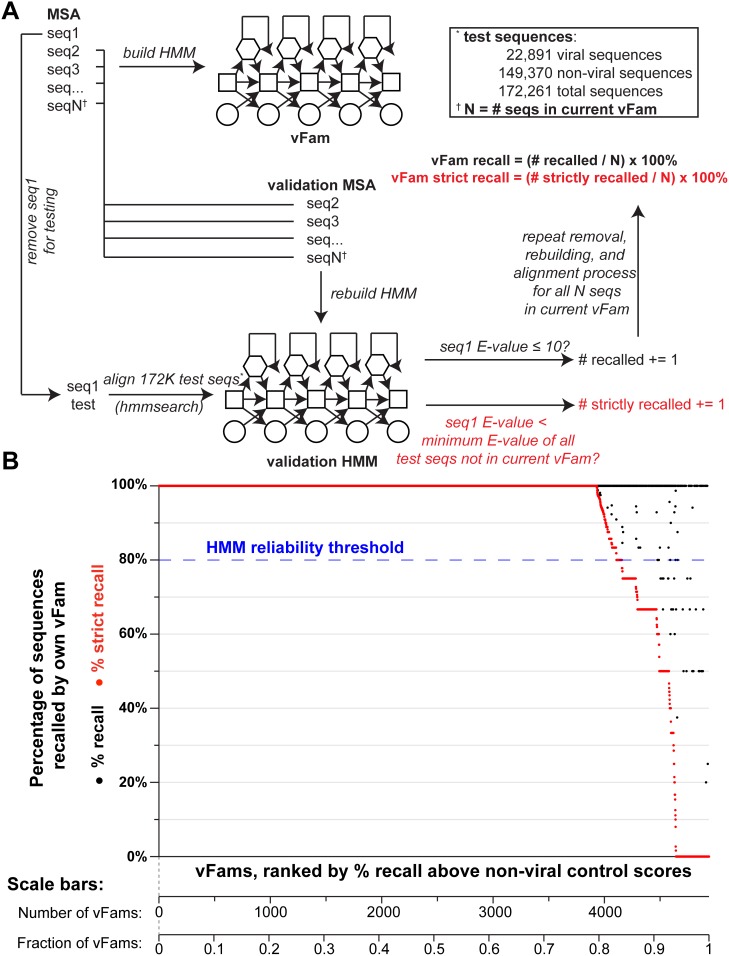
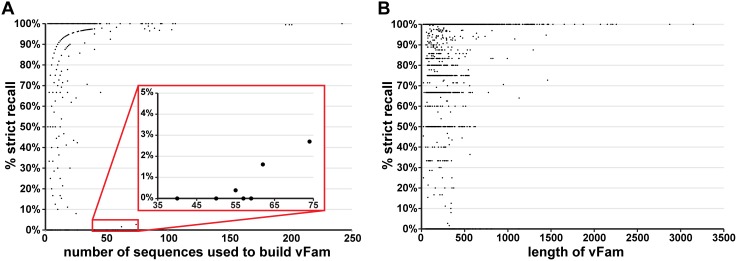
Rapid, sensitive, and specific virus detection is an important component of clinical diagnostics. Massively parallel sequencing enables new diagnostic opportunities that complement traditional serological and PCR based techniques. While massively parallel sequencing promises the benefits of being more comprehensive and less biased than traditional approaches, it presents new analytical challenges, especially with respect to detection of pathogen sequences in metagenomic contexts. To a first approximation, the initial detection of viruses can be achieved simply through alignment of sequence reads or assembled contigs to a reference database of pathogen genomes with tools such as BLAST. However, recognition of highly divergent viral sequences is problematic, and may be further complicated by the inherently high mutation rates of some viral types, especially RNA viruses. In these cases, increased sensitivity may be achieved by leveraging position-specific information during the alignment process. Here, we constructed HMMER3-compatible profile hidden Markov models (profile HMMs) from all the virally annotated proteins in RefSeq in an automated fashion using a custom-built bioinformatic pipeline. We then tested the ability of these viral profile HMMs ("vFams") to accurately classify sequences as viral or non-viral. Cross-validation experiments with full-length gene sequences showed that the vFams were able to recall 91% of left-out viral test sequences without erroneously classifying any non-viral sequences into viral protein clusters. Thorough reanalysis of previously published metagenomic datasets with a set of the best-performing vFams showed that they were more sensitive than BLAST for detecting sequences originating from more distant relatives of known viruses. To facilitate the use of the vFams for rapid detection of remote viral homologs in metagenomic data, we provide two sets of vFams, comprising more than 4,000 vFams each, in the HMMER3 format. We also provide the software necessary to build custom profile HMMs or update the vFams as more viruses are discovered (http://derisilab.ucsf.edu/software/vFam).
快速、灵敏且特异的病毒检测是临床诊断的重要组成部分。大规模平行测序带来了新的诊断机会,可补充传统的血清学和基于PCR的技术。虽然大规模平行测序有望比传统方法更全面且偏差更小,但它也带来了新的分析挑战,特别是在宏基因组背景下检测病原体序列方面。初步估计,病毒的初始检测可通过使用诸如BLAST等工具将序列读数或组装的重叠群与病原体基因组参考数据库进行比对来简单实现。然而,识别高度分化的病毒序列存在问题,并且可能因某些病毒类型(尤其是RNA病毒)固有的高突变率而进一步复杂化。在这些情况下,可通过在比对过程中利用位置特异性信息来提高灵敏度。在此,我们使用定制的生物信息学管道以自动化方式从RefSeq中所有经过病毒注释的蛋白质构建了与HMMER3兼容的轮廓隐马尔可夫模型(轮廓HMM)。然后,我们测试了这些病毒轮廓HMM(“vFams”)将序列准确分类为病毒或非病毒的能力。对全长基因序列进行的交叉验证实验表明,vFams能够召回91%被遗漏的病毒测试序列,且不会将任何非病毒序列错误分类到病毒蛋白簇中。用一组性能最佳的vFams对先前发表的宏基因组数据集进行全面重新分析表明,它们在检测源自已知病毒较远亲属的序列方面比BLAST更灵敏。为便于使用vFams在宏基因组数据中快速检测远距离病毒同源物,我们以HMMER3格式提供了两组vFams,每组包含4000多个vFams。我们还提供了构建定制轮廓HMM或随着发现更多病毒更新vFams所需的软件(http://derisilab.ucsf.edu/software/vFam)。