Kourouklaris Andreas, Ioannou Kyriakos, Athanasiou Ioannis, Panagidou Alexia, Demetriou Kiproulla, Zavros Michalis
Department of Nephrology, Nicosia General Hospital, B1, Strovolos, Nicosia, Cyprus.
J Med Case Rep. 2014 Sep 14;8:307. doi: 10.1186/1752-1947-8-307.
Differential diagnosis of thrombotic microangiopathies can be difficult. Atypical hemolytic uremic syndrome is a rare, life-threatening disease caused by uncontrolled chronic activation of alternative complement pathway, resulting in microvascular thrombosis, organ ischemia and damage. Prognosis is poor: up to 65 percent of patients require dialysis or have kidney damage of varying severity or die despite plasma exchange/plasma infusion treatment.
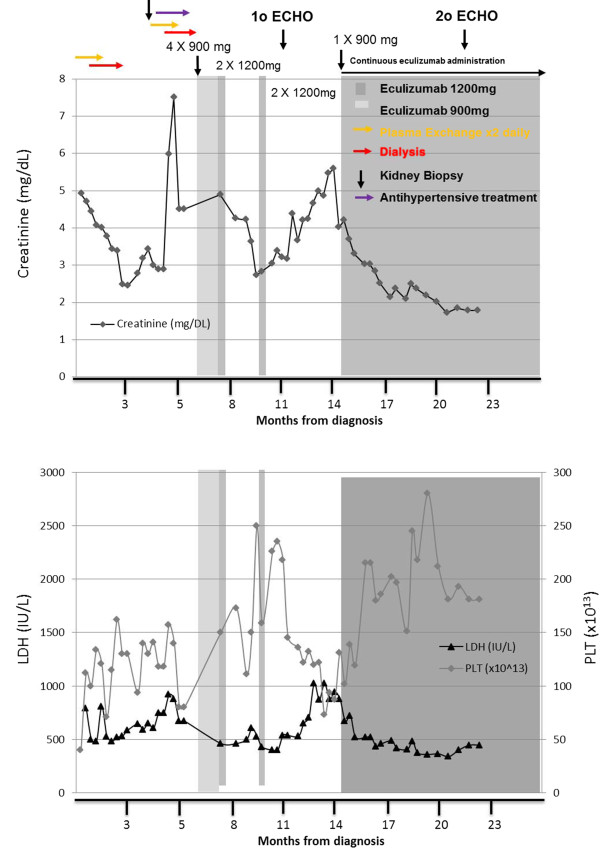
We describe the case of a 23-year-old woman of Hellenic origin who, after a preeclampsia-induced premature delivery, developed thrombotic microangiopathy with renal failure, tonicoclonic seizures, anasarca edema and hypertension. Intensive plasma exchange was initiated twice daily, in parallel to dialysis for one month. Three months later, our patient was discharged with nondialysis-dependent renal failure and without signs of hemolysis. Three months after discharge our patient was readmitted with cardiomyopathy (left ventricular ejection fraction of 25 percent) and signs and symptoms of thrombotic microangiopathy. Our patient was diagnosed with atypical hemolytic uremic syndrome and was started on eculizumab (a complement inhibitor), which improved clinical and laboratory parameters. However, a transient pause in treatment resulted in thrombotic microangiopathy relapse, which was rapidly blocked with reintroduction of eculizumab treatment. During long-term eculizumab treatment, thrombotic microangiopathy manifestations were inhibited and renal and cardiac function restored, with no need for other invasive treatments.
Establishing the diagnosis of atypical hemolytic uremic syndrome in patients presenting with thrombotic microangiopathy is challenging since common symptoms are shared with other conditions like Shiga toxin-producing Escherichia coli hemolytic uremic syndrome and thrombotic thrombocytopenic purpura. The described case illustrates the complexity and importance of rapid diagnosis in a rare disease and the need for appropriate and specific treatment for best long-term outcomes.
血栓性微血管病的鉴别诊断可能具有挑战性。非典型溶血性尿毒症综合征是一种罕见的、危及生命的疾病,由替代补体途径的不受控制的慢性激活引起,导致微血管血栓形成、器官缺血和损伤。预后较差:高达65%的患者需要透析,或有不同程度的肾损伤,或尽管接受了血浆置换/血浆输注治疗仍死亡。
我们描述了一例23岁希腊裔女性的病例,该患者在子痫前期导致的早产之后,出现了伴有肾衰竭、强直性阵挛性癫痫、全身性水肿和高血压的血栓性微血管病。每天进行两次强化血浆置换,同时进行为期一个月的透析。三个月后,我们的患者出院时患有非透析依赖型肾衰竭,且无溶血迹象。出院三个月后,我们的患者因心肌病(左心室射血分数为25%)以及血栓性微血管病的体征和症状再次入院。我们的患者被诊断为非典型溶血性尿毒症综合征,并开始使用依库珠单抗(一种补体抑制剂)治疗,这改善了临床和实验室参数。然而,治疗的短暂中断导致血栓性微血管病复发,重新引入依库珠单抗治疗后迅速得到控制。在长期依库珠单抗治疗期间,血栓性微血管病表现受到抑制,肾和心功能得以恢复,无需其他侵入性治疗。
对于出现血栓性微血管病的患者,确立非典型溶血性尿毒症综合征的诊断具有挑战性,因为其常见症状与其他疾病如产志贺毒素大肠杆菌溶血性尿毒症综合征和血栓性血小板减少性紫癜相同。所描述的病例说明了在罕见疾病中快速诊断的复杂性和重要性,以及为实现最佳长期结果而进行适当和特异性治疗的必要性。