Ely Kenneth H, Matsuoka Mitsuo, DeBerge Matthew P, Ruby Jessica A, Liu Jun, Schneider Mark J, Wang Yan, Hahn Young S, Enelow Richard I
Department of Medicine, Geisel School of Medicine at Dartmouth, Lebanon, New Hampshire, United States of America.
Department of Medicine, Yale University School of Medicine, New Haven, Connecticut, United States of America.
PLoS One. 2014 Sep 24;9(9):e108385. doi: 10.1371/journal.pone.0108385. eCollection 2014.
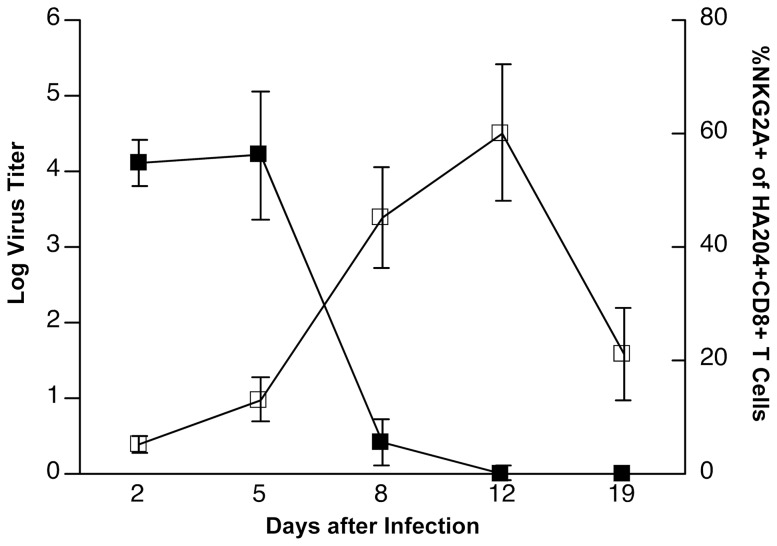
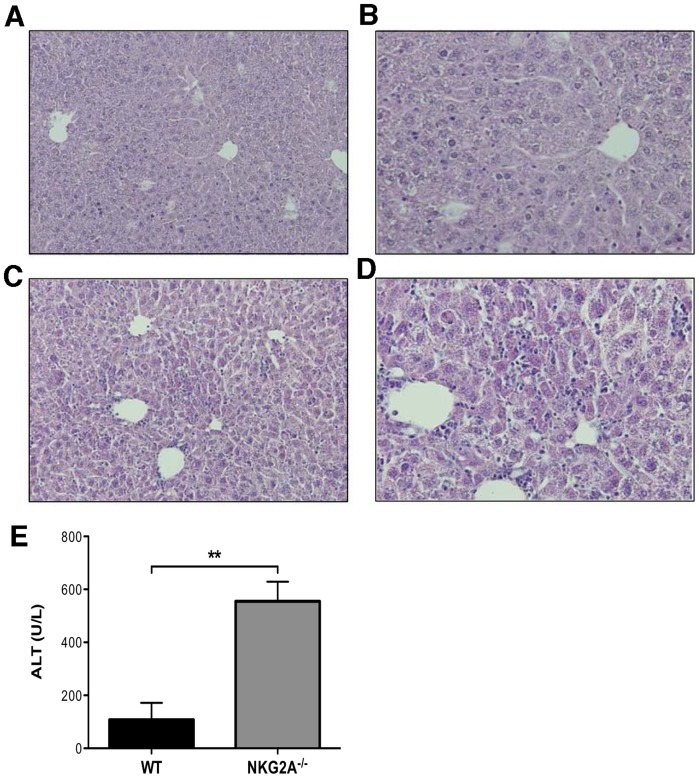
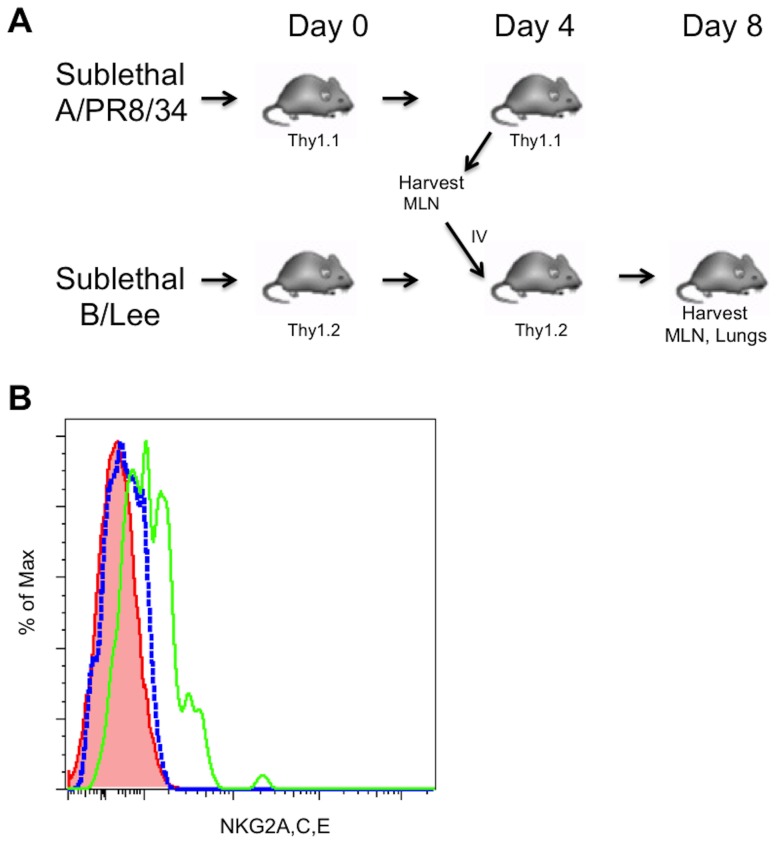
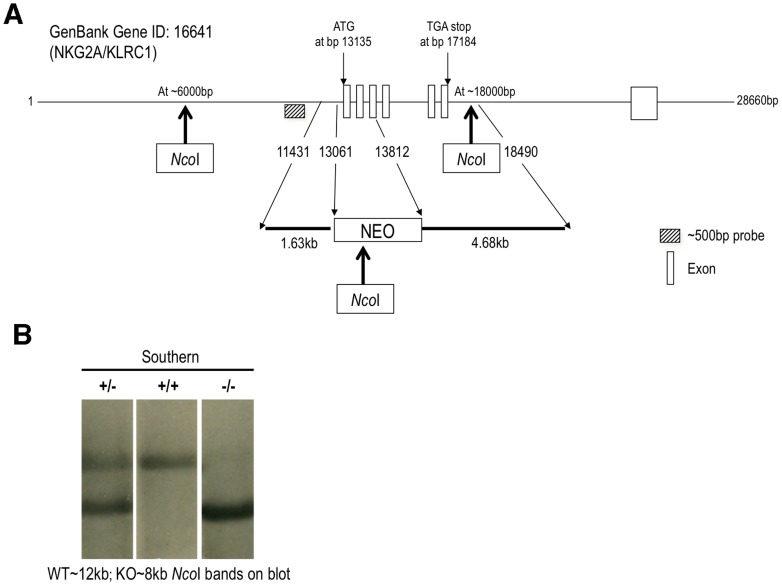
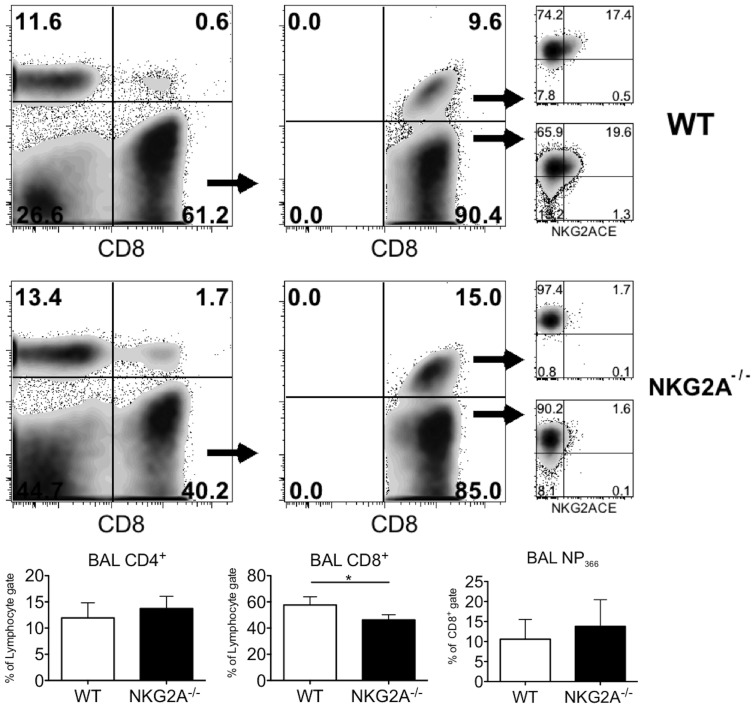
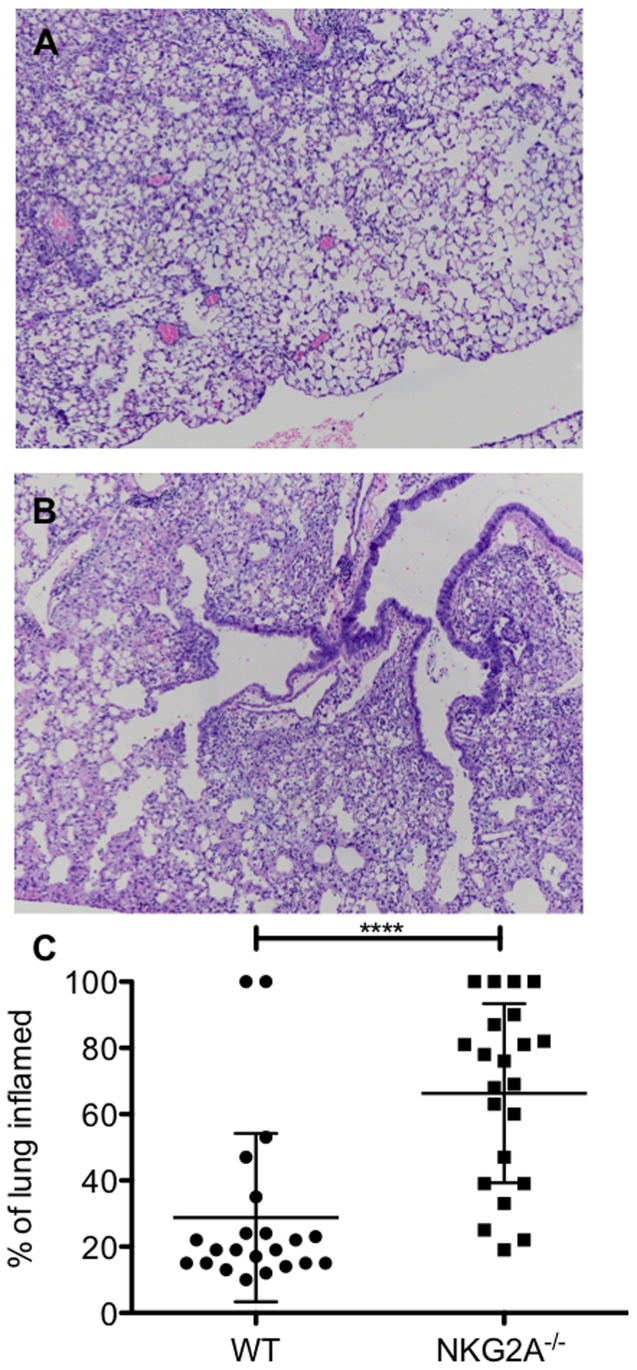
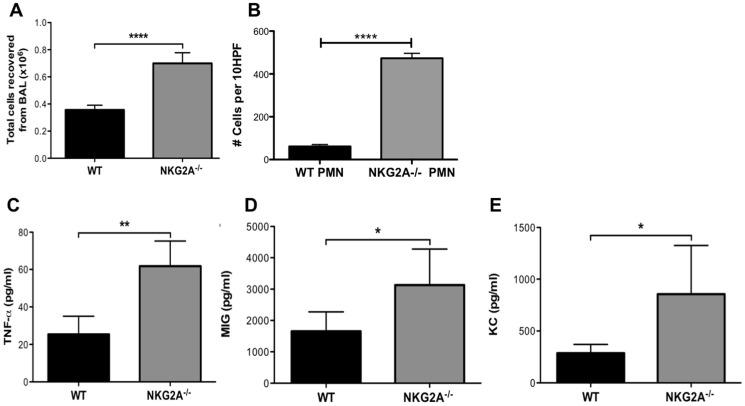
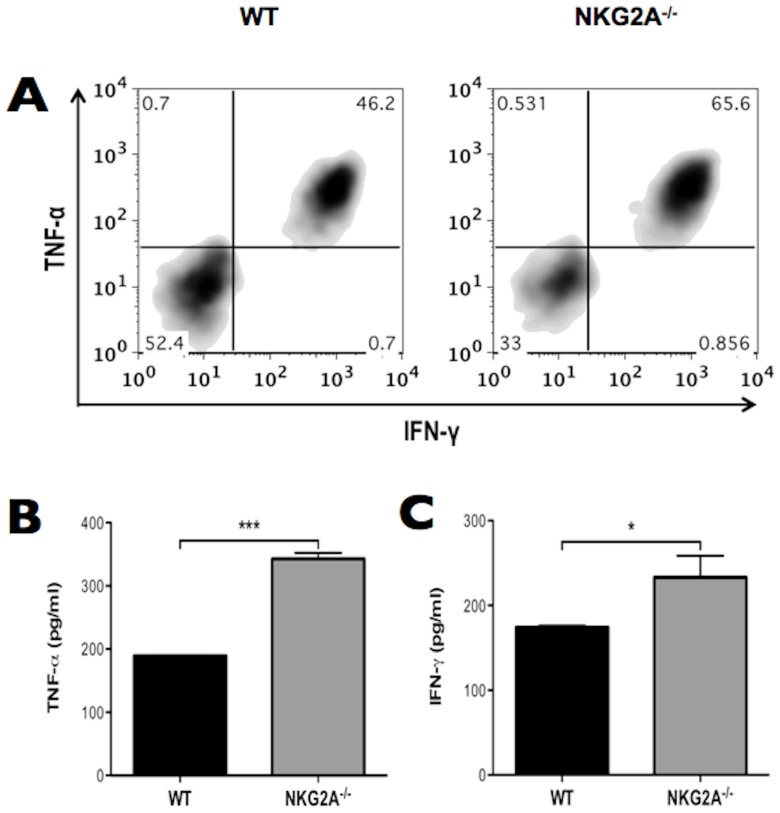
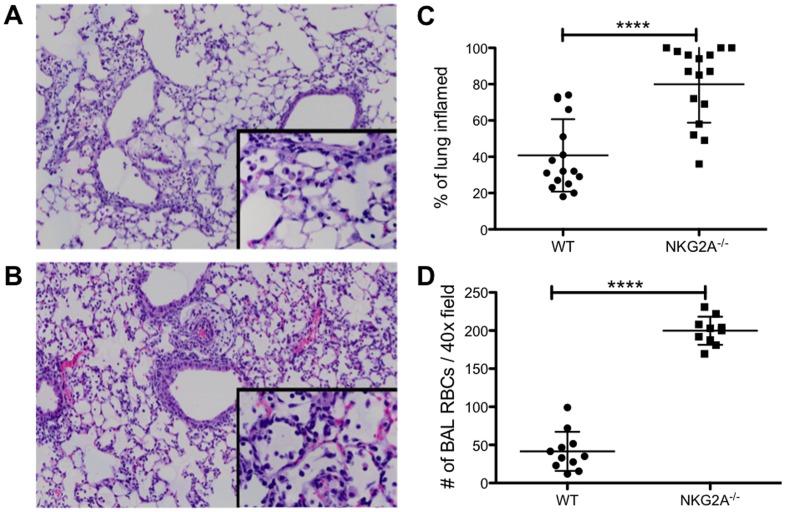
Virus infection triggers a CD8(+) T cell response that aids in virus clearance, but also expresses effector functions that may result in tissue injury. CD8(+) T cells express a variety of activating and inhibiting ligands, though regulation of the expression of inhibitory receptors is not well understood. The ligand for the inhibitory receptor, NKG2A, is the non-classical MHC-I molecule Qa1(b), which may also serve as a putative restricting element for the T cell receptors of purported regulatory CD8(+) T cells. We have previously shown that Qa1(b)-null mice suffer considerably enhanced immunopathologic lung injury in the context of CD8(+) T cell-mediated clearance of influenza infection, as well as evidence in a non-viral system that failure to ligate NKG2A on CD8(+) effector T cells may represent an important component of this process. In this report, we examine the requirements for induction of NKG2A expression, and show that NKG2A expression by CD8(+) T cells occurs as a result of migration from the MLN to the inflammatory lung environment, irrespective of peripheral antigen recognition. Further, we confirmed that NKG2A is a mediator in limiting immunopathology in virus infection using mice with a targeted deletion of NKG2A, and infecting the mutants with two different viruses, influenza and adenovirus. In neither infection is virus clearance altered. In influenza infection, the enhanced lung injury was associated with increased chemoattractant production, increased infiltration of inflammatory cells, and significantly enhanced alveolar hemorrhage. The primary mechanism of enhanced injury was the loss of negative regulation of CD8(+) T cell effector function. A similar effect was observed in the livers of mutant mice infected intravenously with adenovirus. These results demonstrate the immunoregulatory role of CD8(+) NKG2A expression in virus infection, which negatively regulates T cell effector functions and contributes to protection of tissue integrity during virus clearance.
病毒感染引发CD8(+) T细胞反应,这有助于病毒清除,但同时也表达可能导致组织损伤的效应功能。CD8(+) T细胞表达多种激活和抑制配体,不过抑制性受体表达的调控尚不清楚。抑制性受体NKG2A的配体是非经典MHC-I分子Qa1(b),它也可能作为所谓调节性CD8(+) T细胞的T细胞受体的假定限制元件。我们之前已经表明,在CD8(+) T细胞介导的流感感染清除过程中,Qa1(b)基因敲除小鼠的免疫病理性肺损伤显著增强,并且在非病毒系统中有证据表明,CD8(+)效应T细胞上未能连接NKG2A可能是这一过程的重要组成部分。在本报告中,我们研究了诱导NKG2A表达的条件,并表明CD8(+) T细胞中NKG2A的表达是从肠系膜淋巴结迁移到炎症性肺环境的结果,与外周抗原识别无关。此外,我们使用NKG2A靶向缺失的小鼠并用两种不同病毒(流感病毒和腺病毒)感染突变体,证实NKG2A是限制病毒感染中免疫病理的介质。在这两种感染中,病毒清除均未改变。在流感感染中,肺损伤增强与趋化因子产生增加、炎症细胞浸润增加以及肺泡出血显著增强有关。损伤增强的主要机制是CD8(+) T细胞效应功能负调节的丧失。在用腺病毒静脉感染的突变小鼠肝脏中也观察到了类似的效应。这些结果证明了CD8(+) NKG2A表达在病毒感染中的免疫调节作用,它负调节T细胞效应功能,并有助于在病毒清除过程中保护组织完整性。