Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University , 575 Stadium Mall Drive, West Lafayette, Indiana 47907, United States.
J Chem Inf Model. 2014 Oct 27;54(10):2987-95. doi: 10.1021/ci500426q. Epub 2014 Oct 7.
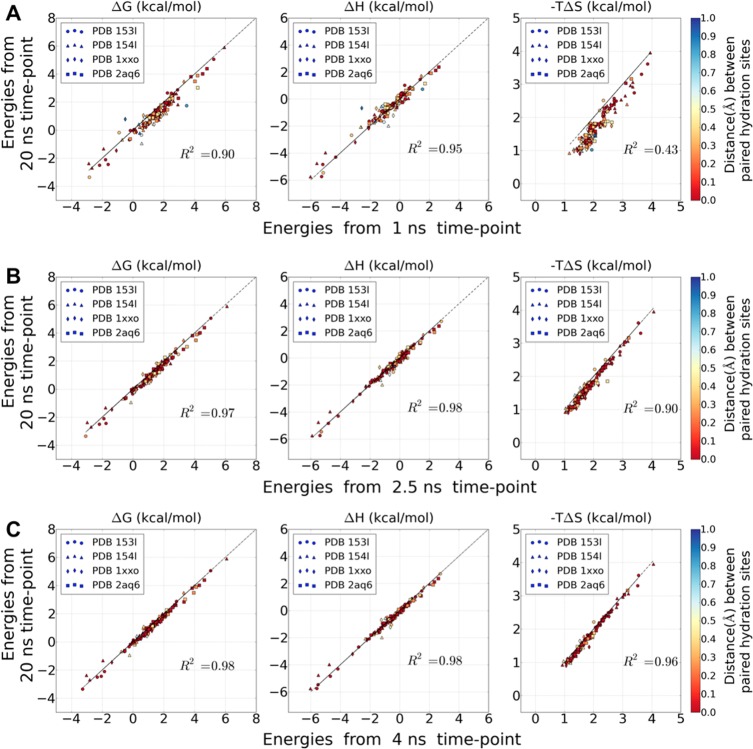
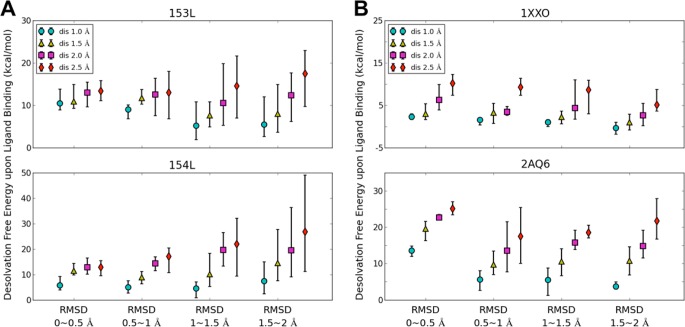
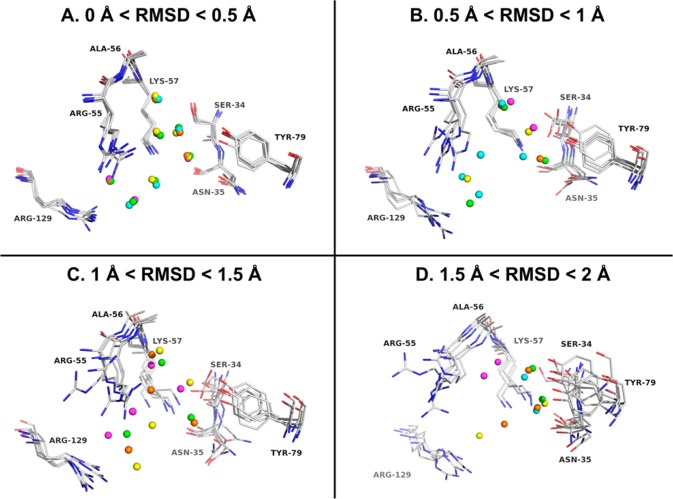
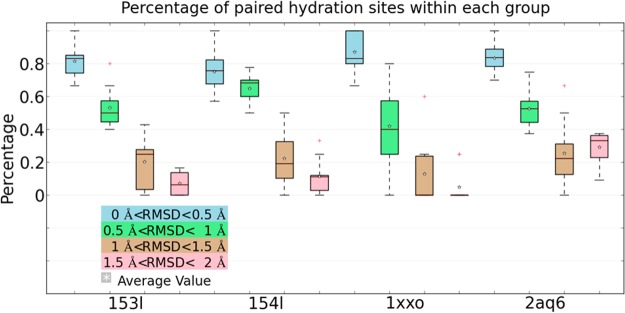
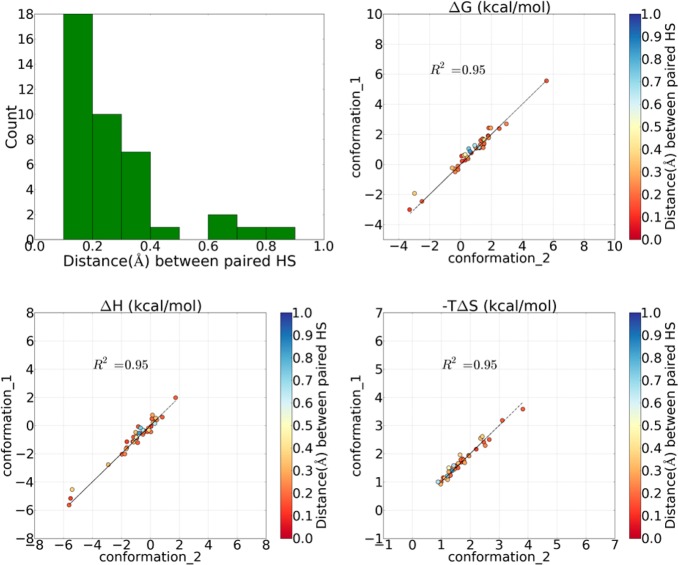
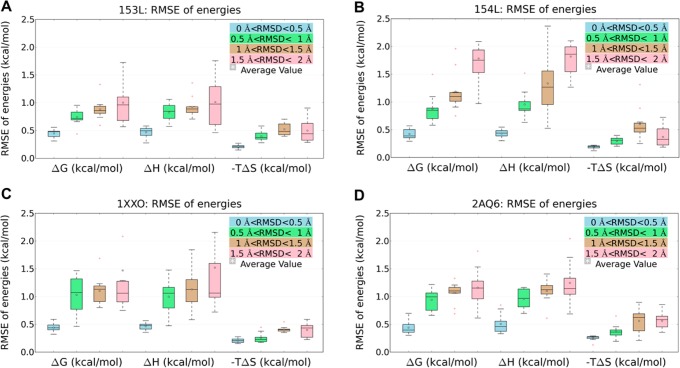
Water contributes significantly to the binding of small molecules to proteins in biochemical systems. Molecular dynamics (MD) simulation based programs such as WaterMap and WATsite have been used to probe the locations and thermodynamic properties of hydration sites at the surface or in the binding site of proteins generating important information for structure-based drug design. However, questions associated with the influence of the simulation protocol on hydration site analysis remain. In this study, we use WATsite to investigate the influence of factors such as simulation length and variations in initial protein conformations on hydration site prediction. We find that 4 ns MD simulation is appropriate to obtain a reliable prediction of the locations and thermodynamic properties of hydration sites. In addition, hydration site prediction can be largely affected by the initial protein conformations used for MD simulations. Here, we provide a first quantification of this effect and further indicate that similar conformations of binding site residues (RMSD < 0.5 Å) are required to obtain consistent hydration site predictions.
水在生化系统中小分子与蛋白质的结合中起着重要作用。基于分子动力学(MD)模拟的程序,如 WaterMap 和 WATsite,已被用于探测蛋白质表面或结合部位的水合位置及其热力学性质,为基于结构的药物设计提供了重要信息。然而,关于模拟方案对水合位置分析影响的问题仍然存在。在这项研究中,我们使用 WATsite 来研究模拟长度和初始蛋白质构象变化等因素对水合位置预测的影响。我们发现,4 ns 的 MD 模拟是获得水合位置的可靠预测的合适长度。此外,水合位置预测可能会受到用于 MD 模拟的初始蛋白质构象的很大影响。在这里,我们首次对这种影响进行了量化,并进一步表明,需要具有相似结合部位残基构象(RMSD <0.5 Å)才能获得一致的水合位置预测。