BMC Bioinformatics. 2014;15 Suppl 9(Suppl 9):S6. doi: 10.1186/1471-2105-15-S9-S6. Epub 2014 Sep 10.
Recent advances in RNA sequencing (RNA-Seq) technology have offered unprecedented scope and resolution for transcriptome analysis. However, precise quantification of mRNA abundance and identification of differentially expressed genes are complicated due to biological and technical variations in RNA-Seq data.
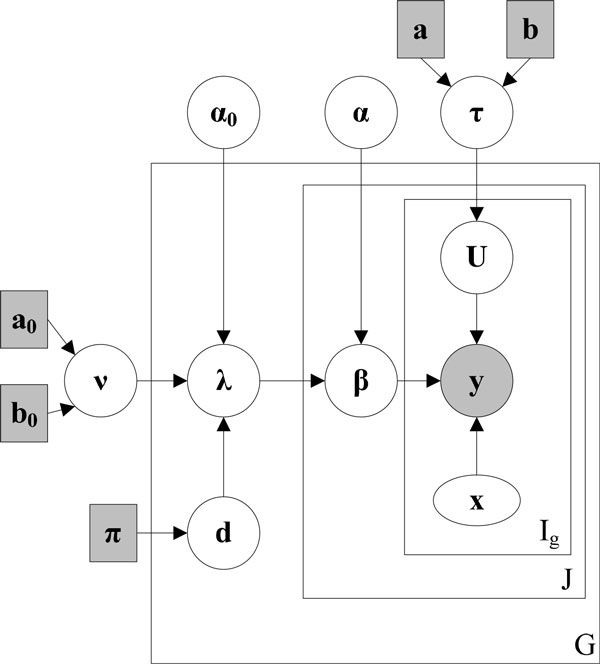
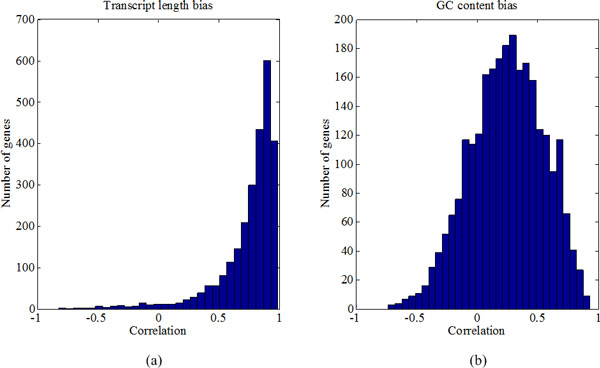
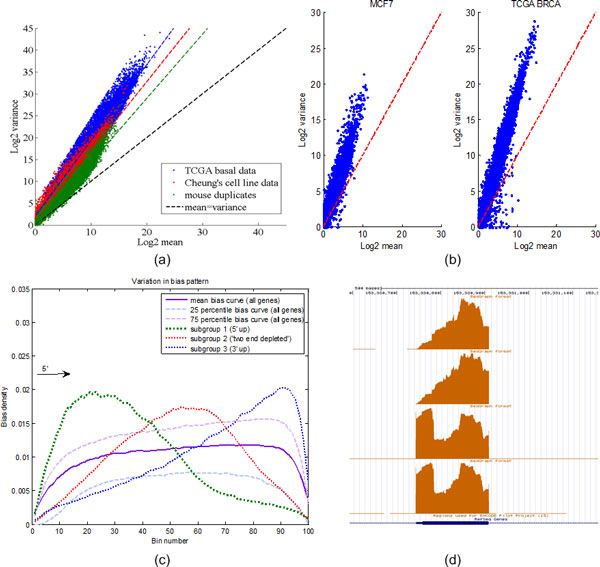
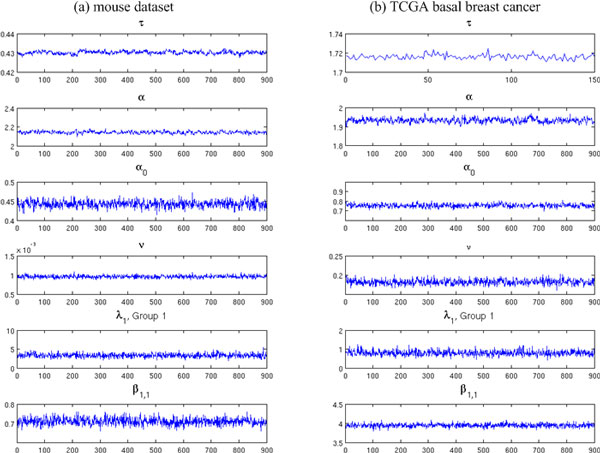
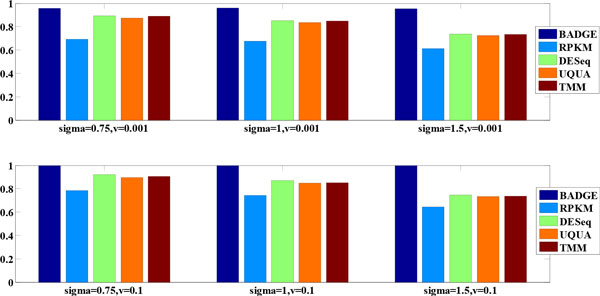
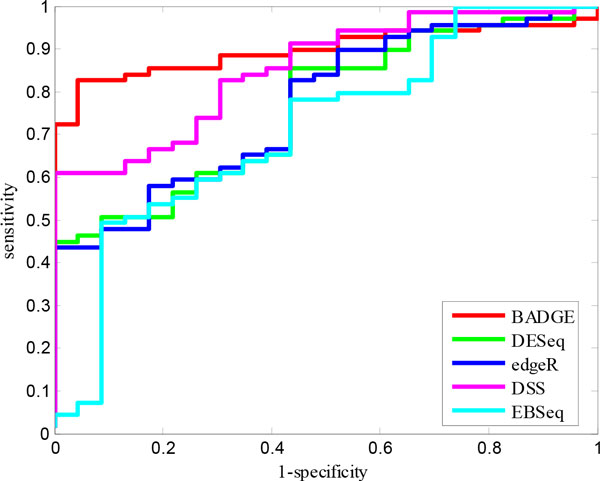
We systematically study the variation in count data and dissect the sources of variation into between-sample variation and within-sample variation. A novel Bayesian framework is developed for joint estimate of gene level mRNA abundance and differential state, which models the intrinsic variability in RNA-Seq to improve the estimation. Specifically, a Poisson-Lognormal model is incorporated into the Bayesian framework to model within-sample variation; a Gamma-Gamma model is then used to model between-sample variation, which accounts for over-dispersion of read counts among multiple samples. Simulation studies, where sequencing counts are synthesized based on parameters learned from real datasets, have demonstrated the advantage of the proposed method in both quantification of mRNA abundance and identification of differentially expressed genes. Moreover, performance comparison on data from the Sequencing Quality Control (SEQC) Project with ERCC spike-in controls has shown that the proposed method outperforms existing RNA-Seq methods in differential analysis. Application on breast cancer dataset has further illustrated that the proposed Bayesian model can 'blindly' estimate sources of variation caused by sequencing biases.
We have developed a novel Bayesian hierarchical approach to investigate within-sample and between-sample variations in RNA-Seq data. Simulation and real data applications have validated desirable performance of the proposed method. The software package is available at http://www.cbil.ece.vt.edu/software.htm.
RNA 测序(RNA-Seq)技术的最新进展为转录组分析提供了前所未有的范围和分辨率。然而,由于 RNA-Seq 数据中的生物学和技术变化,mRNA 丰度的精确定量和差异表达基因的鉴定变得复杂。
我们系统地研究了计数数据的变化,并将变化的来源分解为样品间的变化和样品内的变化。开发了一种新的贝叶斯框架,用于联合估计基因水平的 mRNA 丰度和差异状态,该框架对 RNA-Seq 中的固有变异性进行建模,以改善估计。具体来说,将泊松-对数正态模型纳入贝叶斯框架中以模拟样品内的变化;然后使用伽马-伽马模型来模拟样品间的变化,该模型考虑了多个样品中读取计数的过分散。基于从真实数据集学习到的参数合成测序计数的模拟研究表明,该方法在 mRNA 丰度的定量和差异表达基因的鉴定方面都具有优势。此外,与具有 ERCC Spike-in 对照的测序质量控制(SEQC)项目的数据进行的性能比较表明,该方法在差异分析方面优于现有的 RNA-Seq 方法。在乳腺癌数据集上的应用进一步说明了,所提出的贝叶斯模型可以“盲目”估计测序偏差引起的变异源。
我们开发了一种新的贝叶斯分层方法来研究 RNA-Seq 数据中的样品内和样品间变化。模拟和真实数据应用验证了所提出方法的良好性能。该软件包可在 http://www.cbil.ece.vt.edu/software.htm 获得。