Kuhn Jens H, Andersen Kristian G, Bào Yīmíng, Bavari Sina, Becker Stephan, Bennett Richard S, Bergman Nicholas H, Blinkova Olga, Bradfute Steven, Brister J Rodney, Bukreyev Alexander, Chandran Kartik, Chepurnov Alexander A, Davey Robert A, Dietzgen Ralf G, Doggett Norman A, Dolnik Olga, Dye John M, Enterlein Sven, Fenimore Paul W, Formenty Pierre, Freiberg Alexander N, Garry Robert F, Garza Nicole L, Gire Stephen K, Gonzalez Jean-Paul, Griffiths Anthony, Happi Christian T, Hensley Lisa E, Herbert Andrew S, Hevey Michael C, Hoenen Thomas, Honko Anna N, Ignatyev Georgy M, Jahrling Peter B, Johnson Joshua C, Johnson Karl M, Kindrachuk Jason, Klenk Hans-Dieter, Kobinger Gary, Kochel Tadeusz J, Lackemeyer Matthew G, Lackner Daniel F, Leroy Eric M, Lever Mark S, Mühlberger Elke, Netesov Sergey V, Olinger Gene G, Omilabu Sunday A, Palacios Gustavo, Panchal Rekha G, Park Daniel J, Patterson Jean L, Paweska Janusz T, Peters Clarence J, Pettitt James, Pitt Louise, Radoshitzky Sheli R, Ryabchikova Elena I, Saphire Erica Ollmann, Sabeti Pardis C, Sealfon Rachel, Shestopalov Aleksandr M, Smither Sophie J, Sullivan Nancy J, Swanepoel Robert, Takada Ayato, Towner Jonathan S, van der Groen Guido, Volchkov Viktor E, Volchkova Valentina A, Wahl-Jensen Victoria, Warren Travis K, Warfield Kelly L, Weidmann Manfred, Nichol Stuart T
Integrated Research Facility at Fort Detrick, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Fort Detrick, Frederick, MD 21702, USA.
FAS Center for Systems Biology, Harvard University, Cambridge, MA 02138, USA.
Viruses. 2014 Sep 26;6(9):3663-82. doi: 10.3390/v6093663.
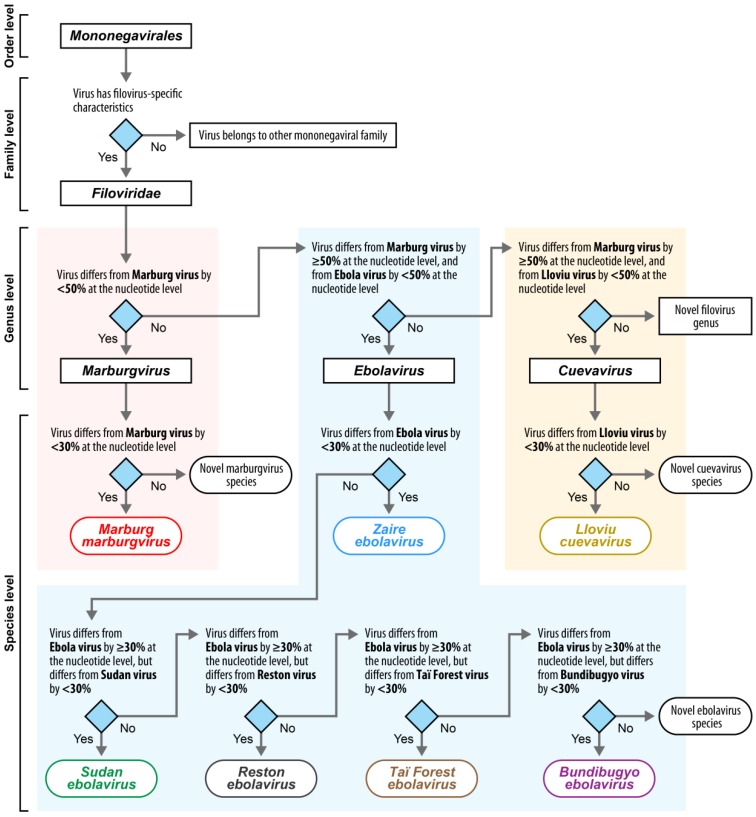
Sequence determination of complete or coding-complete genomes of viruses is becoming common practice for supporting the work of epidemiologists, ecologists, virologists, and taxonomists. Sequencing duration and costs are rapidly decreasing, sequencing hardware is under modification for use by non-experts, and software is constantly being improved to simplify sequence data management and analysis. Thus, analysis of virus disease outbreaks on the molecular level is now feasible, including characterization of the evolution of individual virus populations in single patients over time. The increasing accumulation of sequencing data creates a management problem for the curators of commonly used sequence databases and an entry retrieval problem for end users. Therefore, utilizing the data to their fullest potential will require setting nomenclature and annotation standards for virus isolates and associated genomic sequences. The National Center for Biotechnology Information's (NCBI's) RefSeq is a non-redundant, curated database for reference (or type) nucleotide sequence records that supplies source data to numerous other databases. Building on recently proposed templates for filovirus variant naming [
对病毒的完整基因组或编码完整基因组进行测序,正成为支持流行病学家、生态学家、病毒学家和分类学家工作的常见做法。测序时间和成本正在迅速降低,测序硬件正在改进以供非专业人员使用,软件也在不断完善以简化序列数据管理和分析。因此,现在在分子水平上分析病毒疾病爆发是可行的,包括对单个患者体内单个病毒群体随时间的进化特征进行描述。测序数据的不断积累给常用序列数据库的管理者带来了管理问题,也给终端用户带来了条目检索问题。因此,要充分发挥这些数据的潜力,就需要为病毒分离株和相关基因组序列设定命名和注释标准。美国国立生物技术信息中心(NCBI)的参考序列(RefSeq)是一个用于参考(或类型)核苷酸序列记录的非冗余、经过整理的数据库,它为许多其他数据库提供源数据。基于最近提出的丝状病毒变种命名模板[<病毒名称>(<毒株>)/<分离宿主后缀>/<采样国家>/<采样年份>/<基因变种指定>-<分离株指定>],我们报告了大多数过去和当前活跃的丝状病毒专家就RefSeq中要呈现的8种丝状病毒类型变种和分离株达成的共识决定、它们的最终命名以及它们的相关序列。