Loughrey David, Watters Kyle E, Settle Alexander H, Lucks Julius B
School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, NY 14850, USA.
School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, NY 14850, USA
Nucleic Acids Res. 2014 Dec 1;42(21):e165. doi: 10.1093/nar/gku909. Epub 2014 Oct 10.
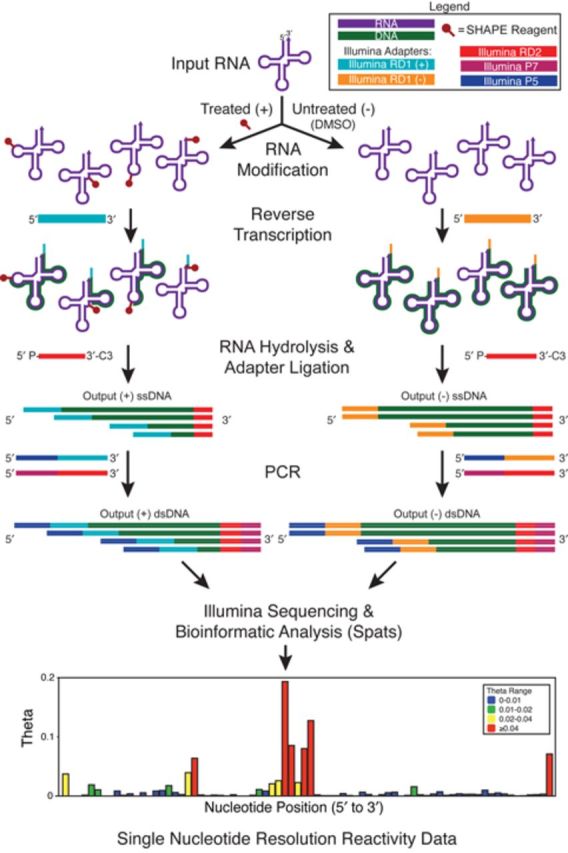
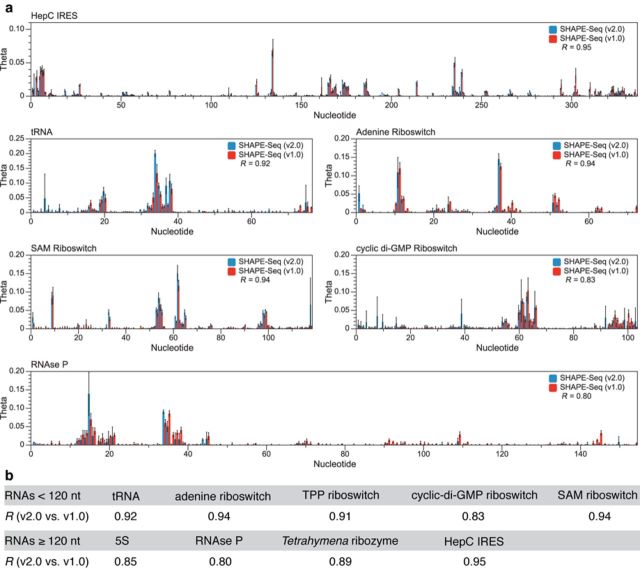
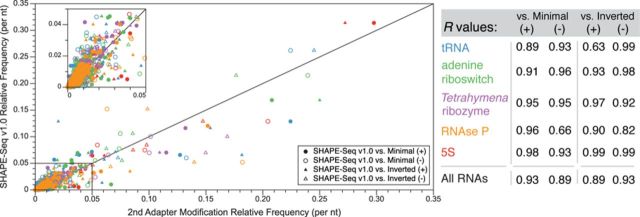
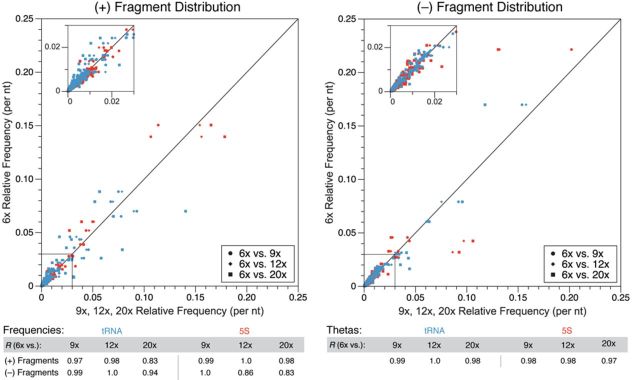
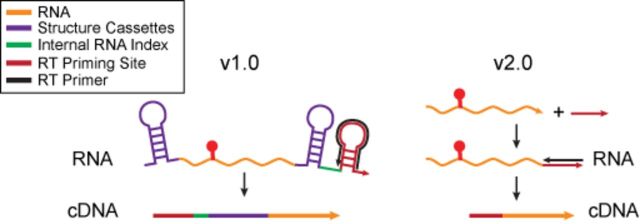
RNA structure is a primary determinant of its function, and methods that merge chemical probing with next generation sequencing have created breakthroughs in the throughput and scale of RNA structure characterization. However, little work has been done to examine the effects of library preparation and sequencing on the measured chemical probe reactivities that encode RNA structural information. Here, we present the first analysis and optimization of these effects for selective 2'-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). We first optimize SHAPE-Seq, and show that it provides highly reproducible reactivity data over a wide range of RNA structural contexts with no apparent biases. As part of this optimization, we present SHAPE-Seq v2.0, a 'universal' method that can obtain reactivity information for every nucleotide of an RNA without having to use or introduce a specific reverse transcriptase priming site within the RNA. We show that SHAPE-Seq v2.0 is highly reproducible, with reactivity data that can be used as constraints in RNA folding algorithms to predict structures on par with those generated using data from other SHAPE methods. We anticipate SHAPE-Seq v2.0 to be broadly applicable to understanding the RNA sequence-structure relationship at the heart of some of life's most fundamental processes.
RNA结构是其功能的主要决定因素,将化学探针与下一代测序相结合的方法在RNA结构表征的通量和规模方面取得了突破。然而,在研究文库制备和测序对编码RNA结构信息的化学探针反应性测量的影响方面,所做的工作很少。在此,我们首次对通过引物延伸测序分析的选择性2'-羟基酰化(SHAPE-Seq)的这些影响进行了分析和优化。我们首先优化了SHAPE-Seq,并表明它在广泛的RNA结构背景下提供了高度可重复的反应性数据,且没有明显偏差。作为该优化的一部分,我们提出了SHAPE-Seq v2.0,这是一种“通用”方法,无需在RNA中使用或引入特定的逆转录酶引物位点,就能获得RNA每个核苷酸的反应性信息。我们表明,SHAPE-Seq v2.0具有高度可重复性,其反应性数据可作为RNA折叠算法中的约束条件,用于预测与使用其他SHAPE方法生成的结构相当的结构。我们预计SHAPE-Seq v2.0将广泛应用于理解一些生命最基本过程核心的RNA序列-结构关系。