Popescu Andrei-Alin, Harper Andrea L, Trick Martin, Bancroft Ian, Huber Katharina T
School of Computing Sciences, University of East Anglia, Norwich Research Park, Norwich, Norfolk NR4 7TJ, United Kingdom.
Centre for Novel Agricultural Products, Department of Biology, University of York, York YO10 5DD, United Kingdom.
Genetics. 2014 Dec;198(4):1421-31. doi: 10.1534/genetics.114.171314. Epub 2014 Oct 16.
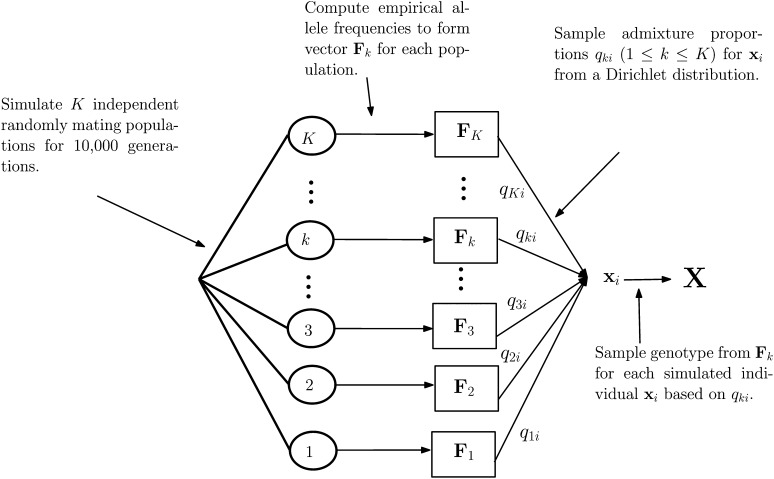
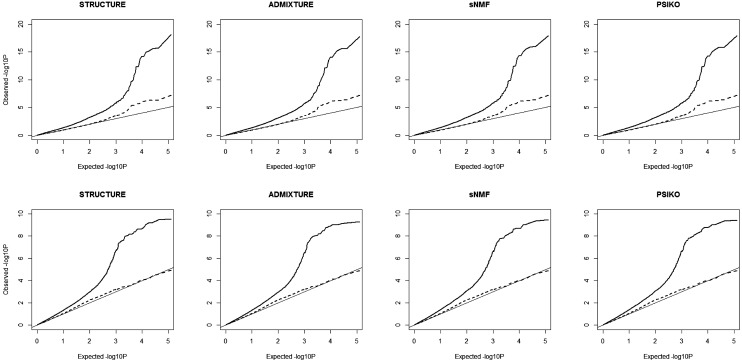
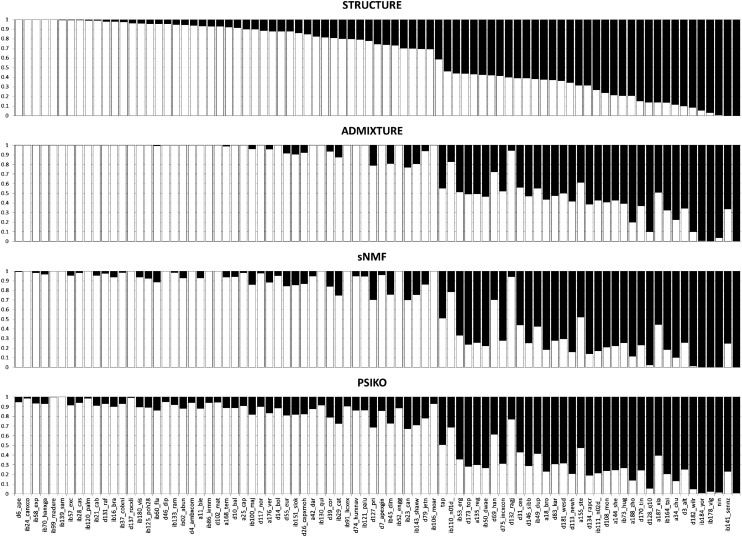
Population structure is a confounding factor in genome-wide association studies, increasing the rate of false positive associations. To correct for it, several model-based algorithms such as ADMIXTURE and STRUCTURE have been proposed. These tend to suffer from the fact that they have a considerable computational burden, limiting their applicability when used with large datasets, such as those produced by next generation sequencing techniques. To address this, nonmodel based approaches such as sparse nonnegative matrix factorization (sNMF) and EIGENSTRAT have been proposed, which scale better with larger data. Here we present a novel nonmodel-based approach, population structure inference using kernel-PCA and optimization (PSIKO), which is based on a unique combination of linear kernel-PCA and least-squares optimization and allows for the inference of admixture coefficients, principal components, and number of founder populations of a dataset. PSIKO has been compared against existing leading methods on a variety of simulation scenarios, as well as on real biological data. We found that in addition to producing results of the same quality as other tested methods, PSIKO scales extremely well with dataset size, being considerably (up to 30 times) faster for longer sequences than even state-of-the-art methods such as sNMF. PSIKO and accompanying manual are freely available at https://www.uea.ac.uk/computing/psiko.
群体结构是全基因组关联研究中的一个混杂因素,会增加假阳性关联的发生率。为了对此进行校正,已经提出了几种基于模型的算法,如ADMIXTURE和STRUCTURE。然而,这些算法往往存在计算负担较重的问题,限制了它们在处理大型数据集(如下一代测序技术产生的数据集)时的适用性。为了解决这个问题,人们又提出了诸如稀疏非负矩阵分解(sNMF)和EIGENSTRAT等基于非模型的方法,这些方法在处理更大数据时扩展性更好。在此,我们提出了一种新颖的基于非模型的方法——使用核主成分分析和优化进行群体结构推断(PSIKO),该方法基于线性核主成分分析和最小二乘法优化的独特组合,能够推断数据集的混合系数、主成分以及奠基群体的数量。我们在各种模拟场景以及真实生物学数据上,将PSIKO与现有的领先方法进行了比较。我们发现,PSIKO除了能产生与其他测试方法质量相当的结果外,在数据集规模方面扩展性极佳,对于较长序列,其速度比诸如sNMF这样的前沿方法快得多(快达30倍)。可在https://www.uea.ac.uk/computing/psiko上免费获取PSIKO及其配套手册。