National Center for Biotechnology Information, National Institutes of Health, Department of Health and Human Services, Bethesda, Maryland 20894 and
Cancer Data Science Laboratory, National Cancer Institute, National Institutes of Health; Department of Health and Human Services; Bethesda, Maryland 20892.
G3 (Bethesda). 2019 Aug 8;9(8):2447-2461. doi: 10.1534/g3.118.200925.
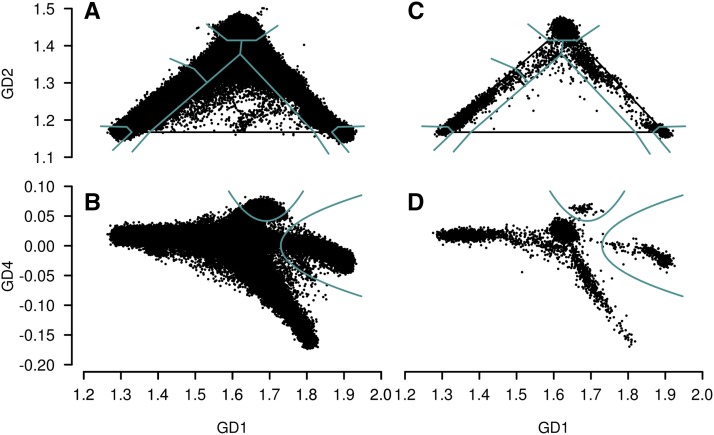
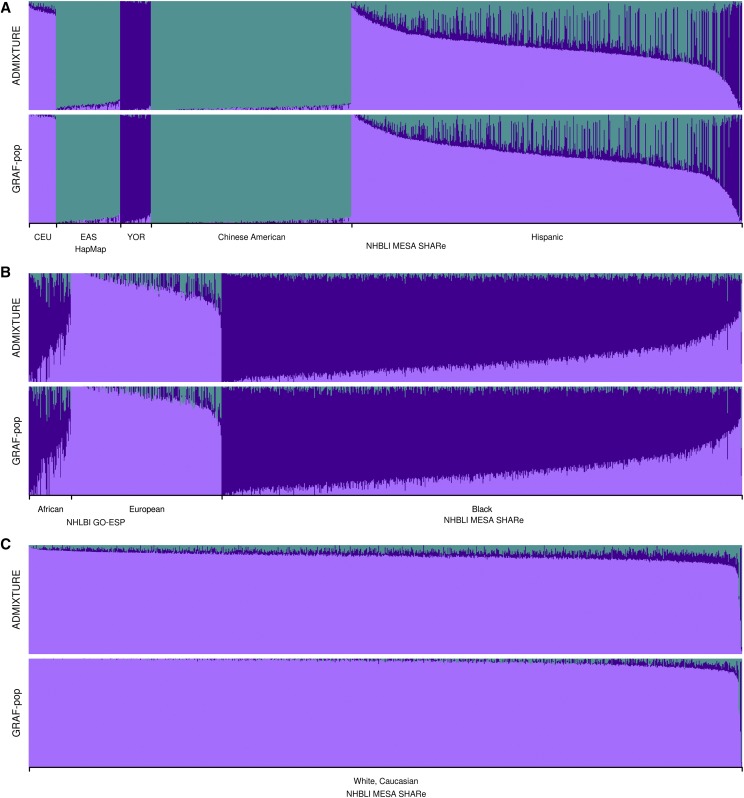
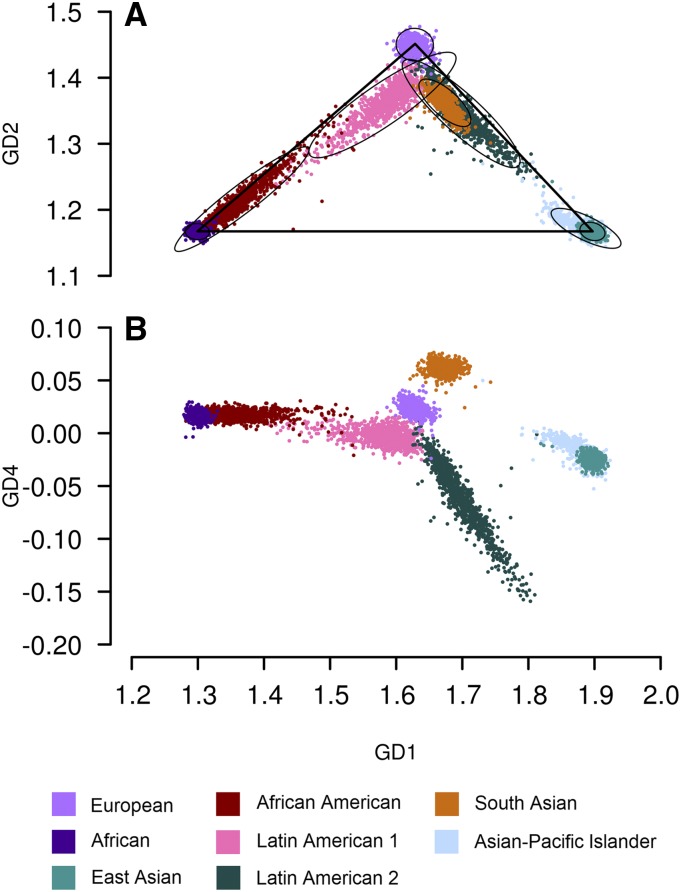
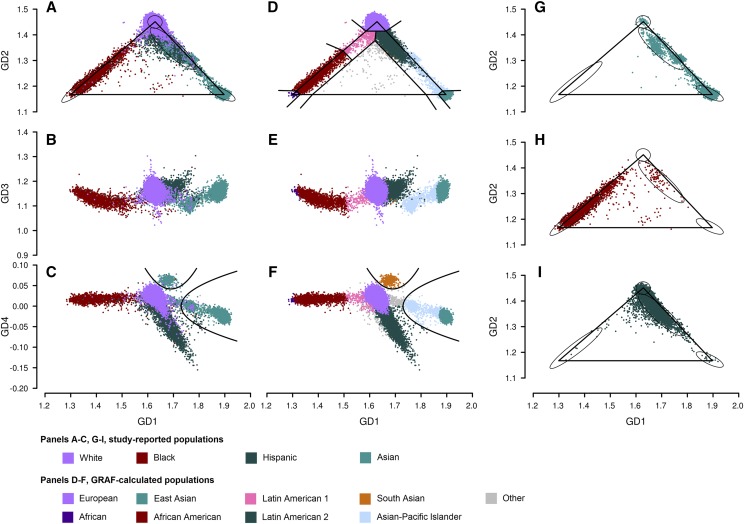
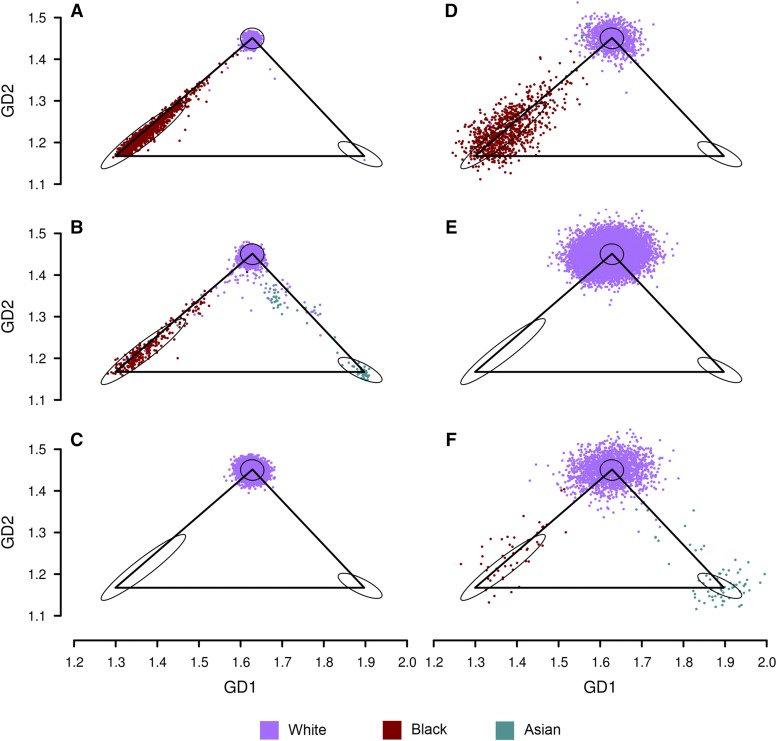
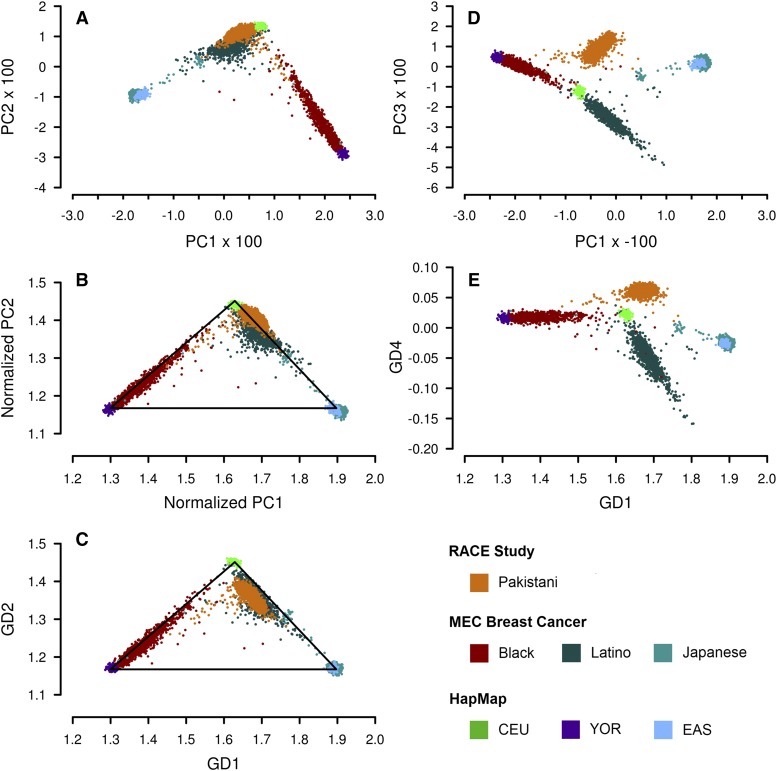
Inferring subject ancestry using genetic data is an important step in genetic association studies, required for dealing with population stratification. It has become more challenging to infer subject ancestry quickly and accurately since large amounts of genotype data, collected from millions of subjects by thousands of studies using different methods, are accessible to researchers from repositories such as the database of Genotypes and Phenotypes (dbGaP) at the National Center for Biotechnology Information (NCBI). Study-reported populations submitted to dbGaP are often not harmonized across studies or may be missing. Widely-used methods for ancestry prediction assume that most markers are genotyped in all subjects, but this assumption is unrealistic if one wants to combine studies that used different genotyping platforms. To provide ancestry inference and visualization across studies, we developed a new method, GRAF-pop, of ancestry prediction that is robust to missing genotypes and allows researchers to visualize predicted population structure in color and in three dimensions. When genotypes are dense, GRAF-pop is comparable in quality and running time to existing ancestry inference methods EIGENSTRAT, FastPCA, and FlashPCA2, all of which rely on principal components analysis (PCA). When genotypes are not dense, GRAF-pop gives much better ancestry predictions than the PCA-based methods. GRAF-pop employs basic geometric and probabilistic methods; the visualized ancestry predictions have a natural geometric interpretation, which is lacking in PCA-based methods. Since February 2018, GRAF-pop has been successfully incorporated into the dbGaP quality control process to identify inconsistencies between study-reported and computationally predicted populations and to provide harmonized population values in all new dbGaP submissions amenable to population prediction, based on marker genotypes. Plots, produced by GRAF-pop, of summary population predictions are available on dbGaP study pages, and the software, is available at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/Software.cgi.
使用遗传数据推断个体的祖先族群是遗传关联研究中的重要步骤,这对于处理群体分层至关重要。由于可以从 NCBI 的 dbGaP 等存储库中获取来自成千上万项研究的数以百万计的个体的大量基因型数据,因此快速准确地推断个体的祖先族群变得更加具有挑战性。向 dbGaP 提交的研究报告的族群通常在不同的研究中没有协调一致,或者可能缺失。广泛使用的祖先预测方法假设大多数标记都在所有个体中进行了基因分型,但如果要合并使用不同基因分型平台的研究,这种假设就不切实际。为了在研究之间提供祖先推断和可视化,我们开发了一种新的祖先预测方法 GRAF-pop,该方法对缺失基因型具有鲁棒性,允许研究人员以颜色和三维形式可视化预测的群体结构。当基因型密集时,GRAF-pop 在质量和运行时间上与现有的祖先推断方法 EIGENSTRAT、FastPCA 和 FlashPCA2 相当,所有这些方法都依赖于主成分分析(PCA)。当基因型不密集时,GRAF-pop 比基于 PCA 的方法给出了更好的祖先预测。GRAF-pop 采用了基本的几何和概率方法;可视化的祖先预测具有自然的几何解释,而基于 PCA 的方法则缺乏这种解释。自 2018 年 2 月以来,GRAF-pop 已成功纳入 dbGaP 质量控制流程,以识别研究报告的族群与计算预测的族群之间的不一致,并根据标记基因型为所有新的可进行族群预测的 dbGaP 提交提供协调一致的族群值。基于 GRAF-pop 生成的总结族群预测图可在 dbGaP 研究页面上查看,该软件可在 https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/Software.cgi 上获得。