Leonard Antony, Paton Adrienne W, El-Quadi Monaliza, Paton James C, Fazal Fabeha
Department of Pediatrics, Lung Biology and Disease Program, University of Rochester School of Medicine and Dentistry, Rochester, New York, United States of America.
Research Centre for Infectious Diseases, School of Molecular and Biomedical Science, University of Adelaide, Adelaide, South Australia, Australia.
PLoS One. 2014 Oct 30;9(10):e110949. doi: 10.1371/journal.pone.0110949. eCollection 2014.
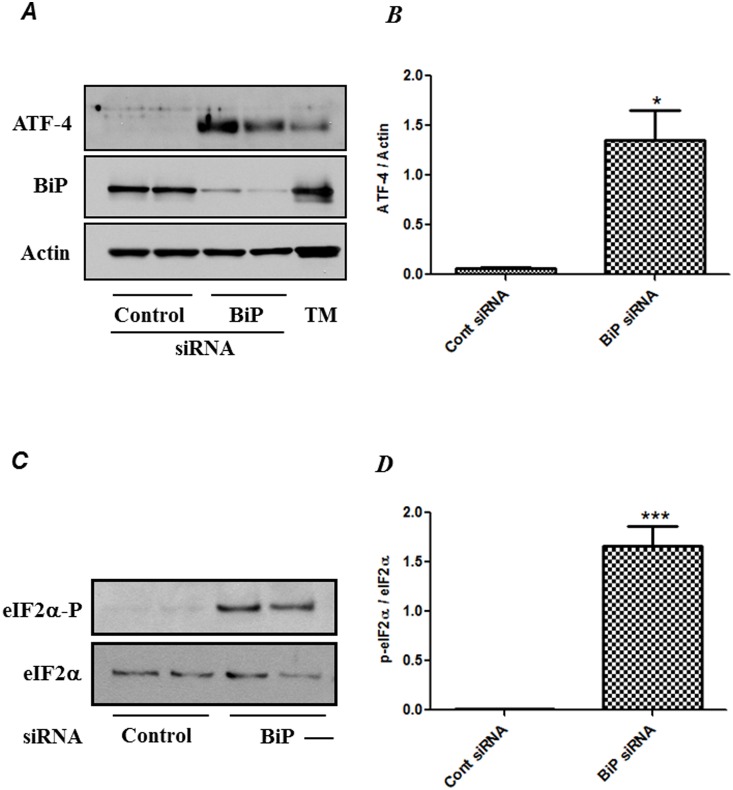
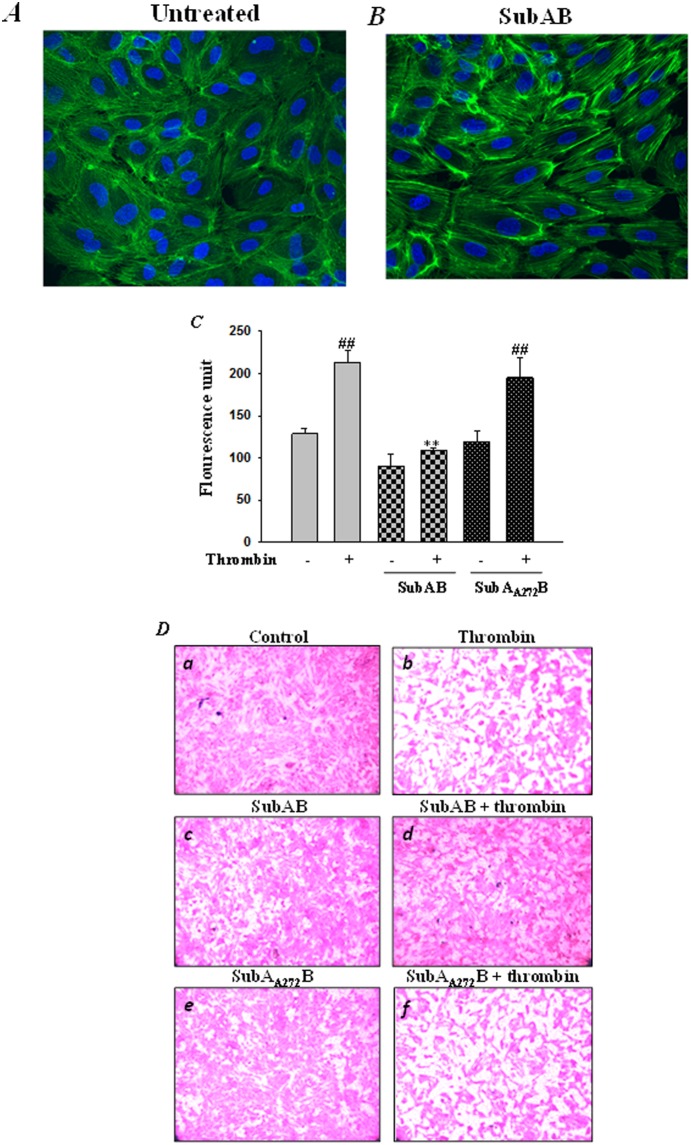
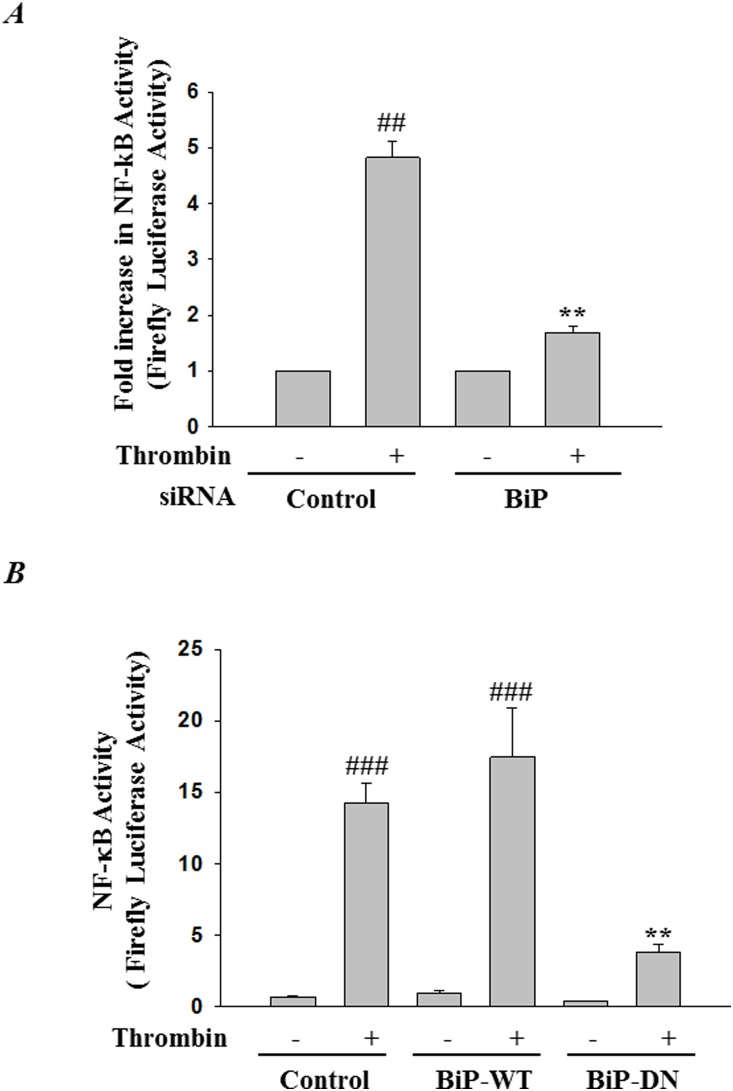
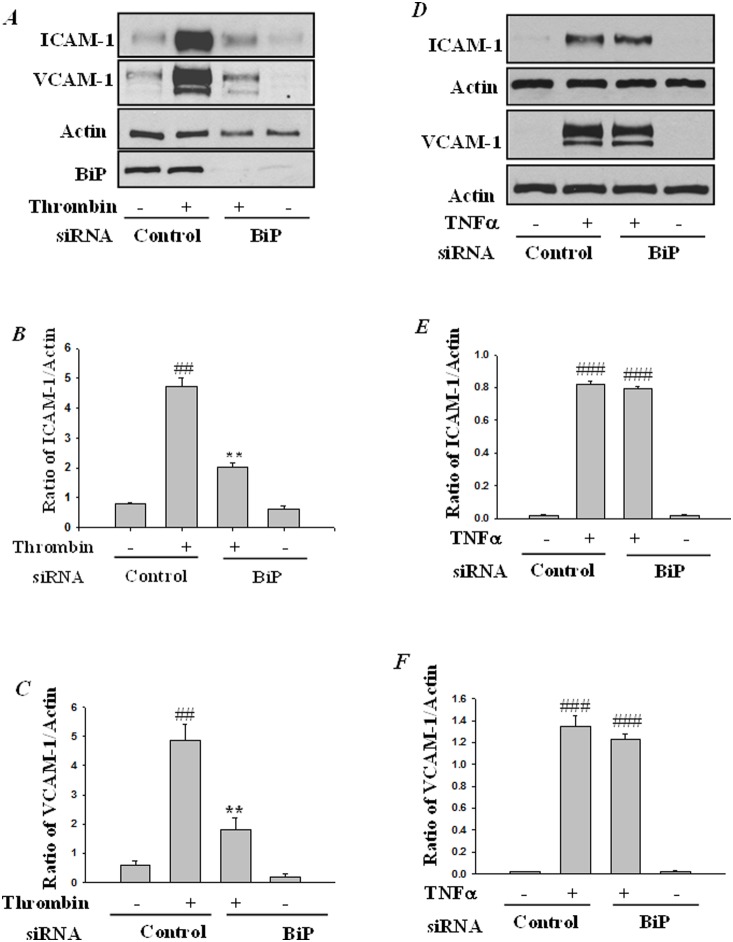
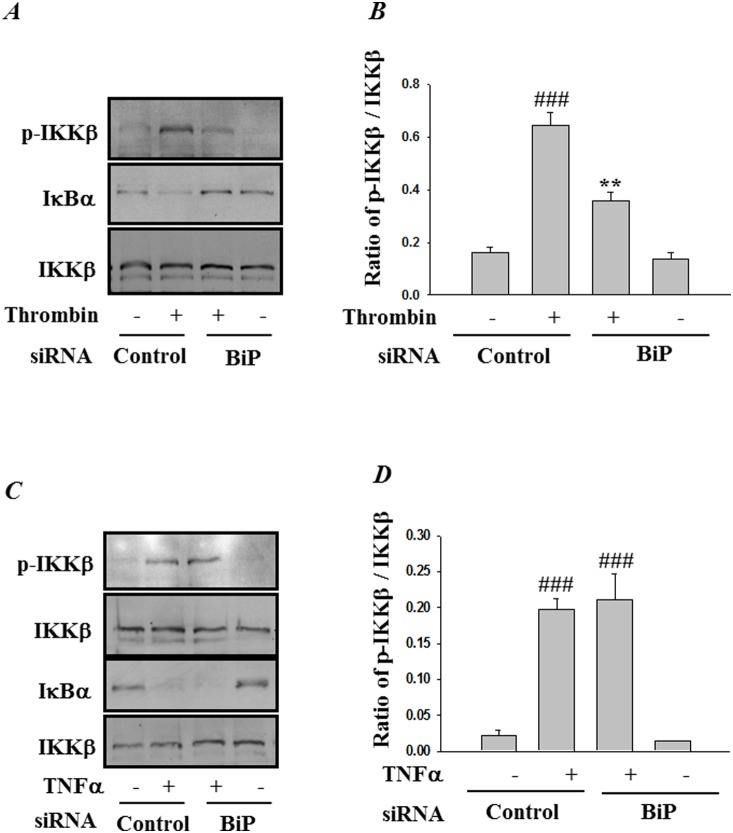
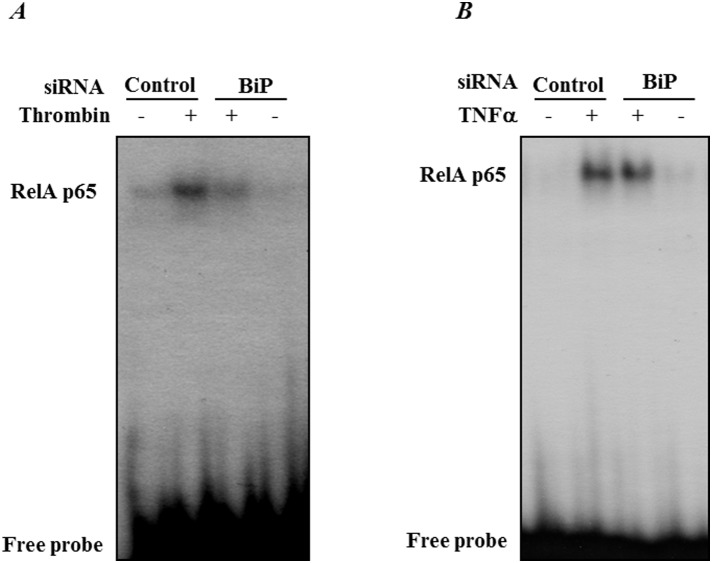
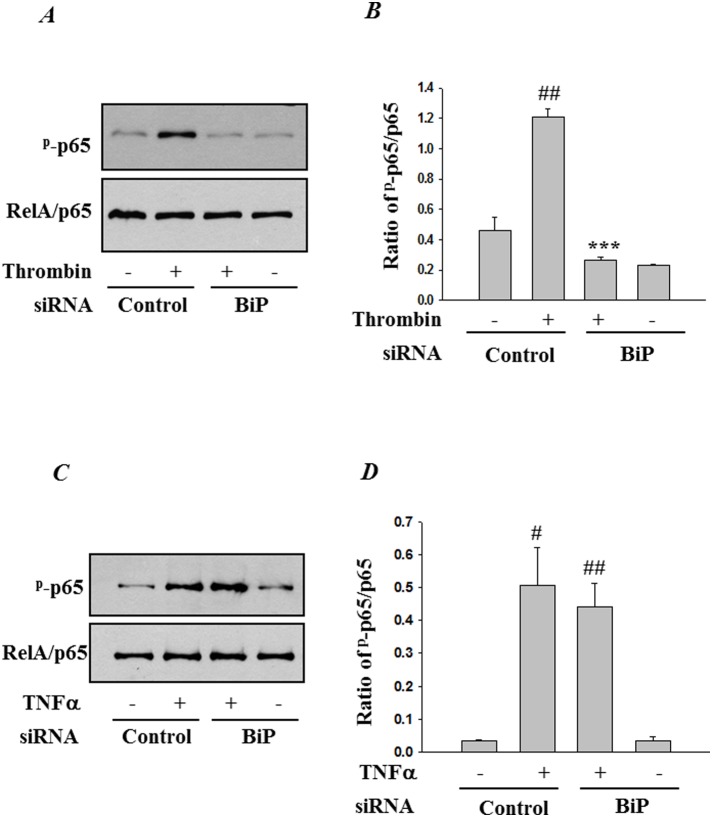
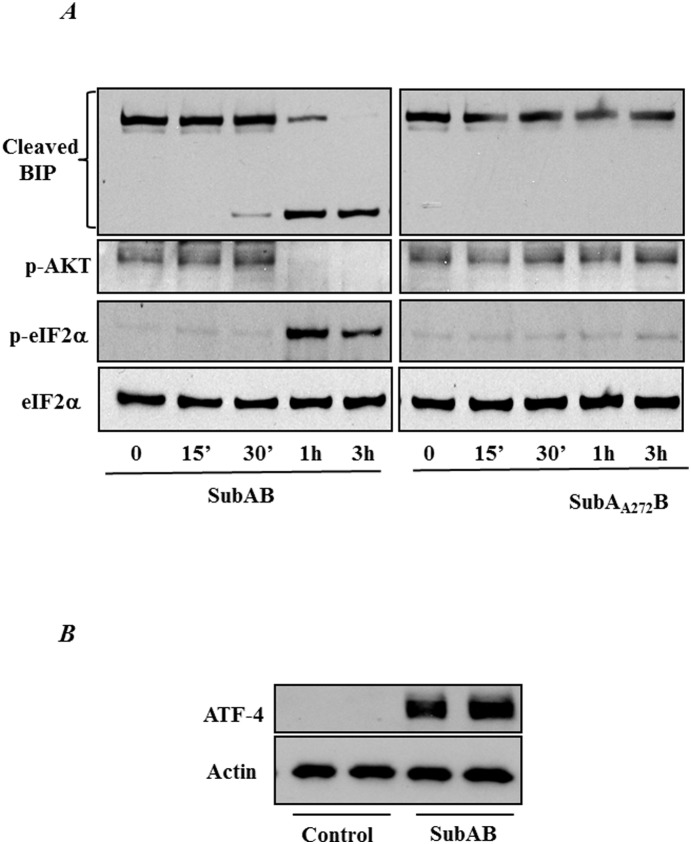
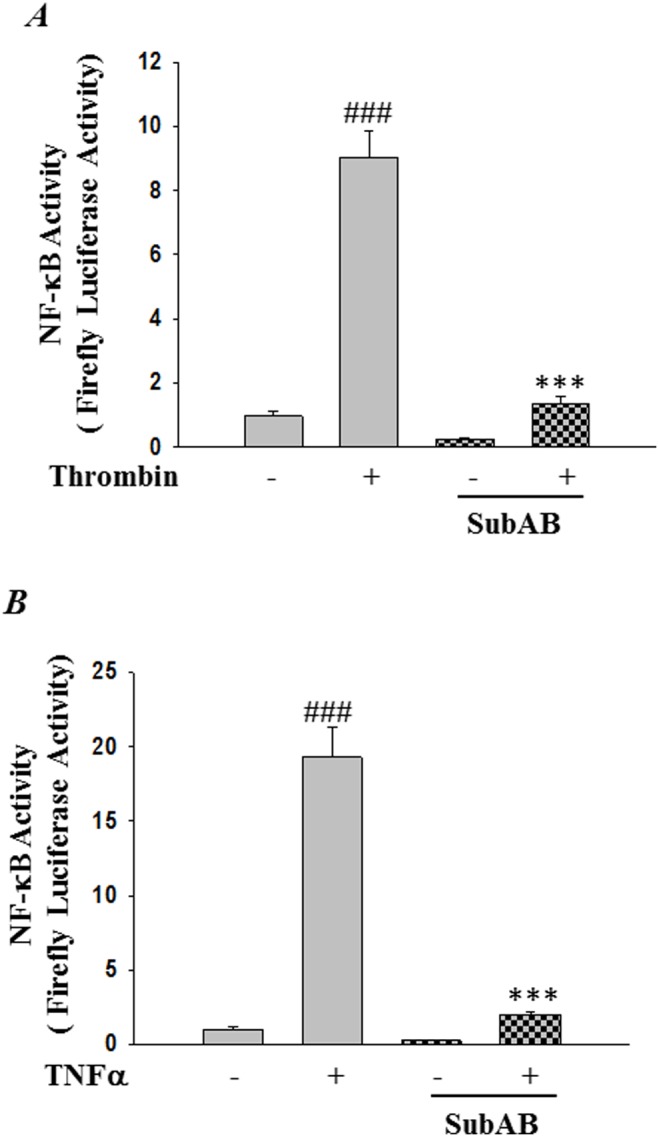
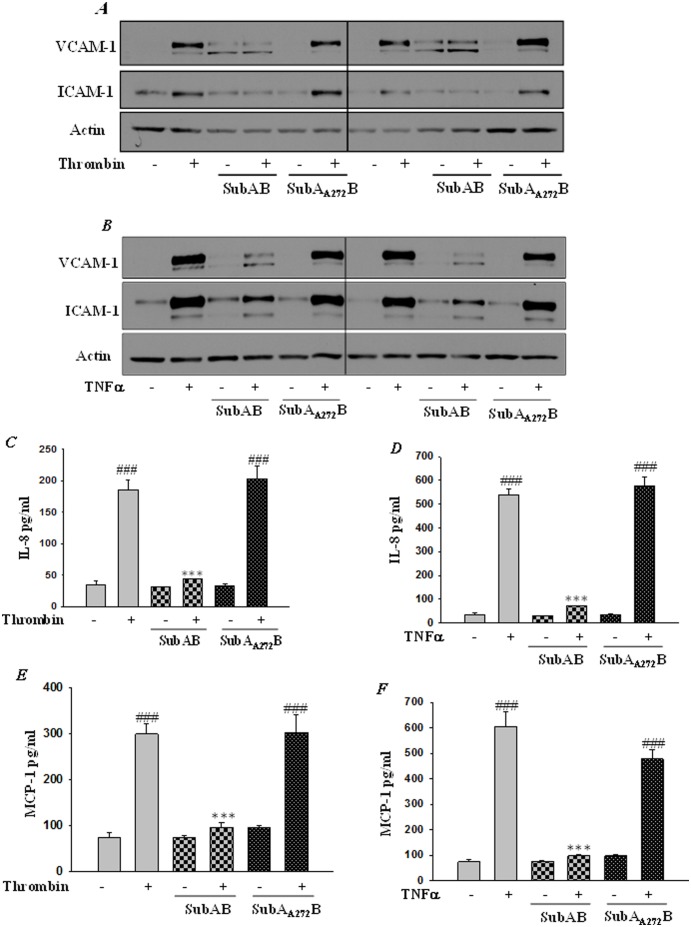
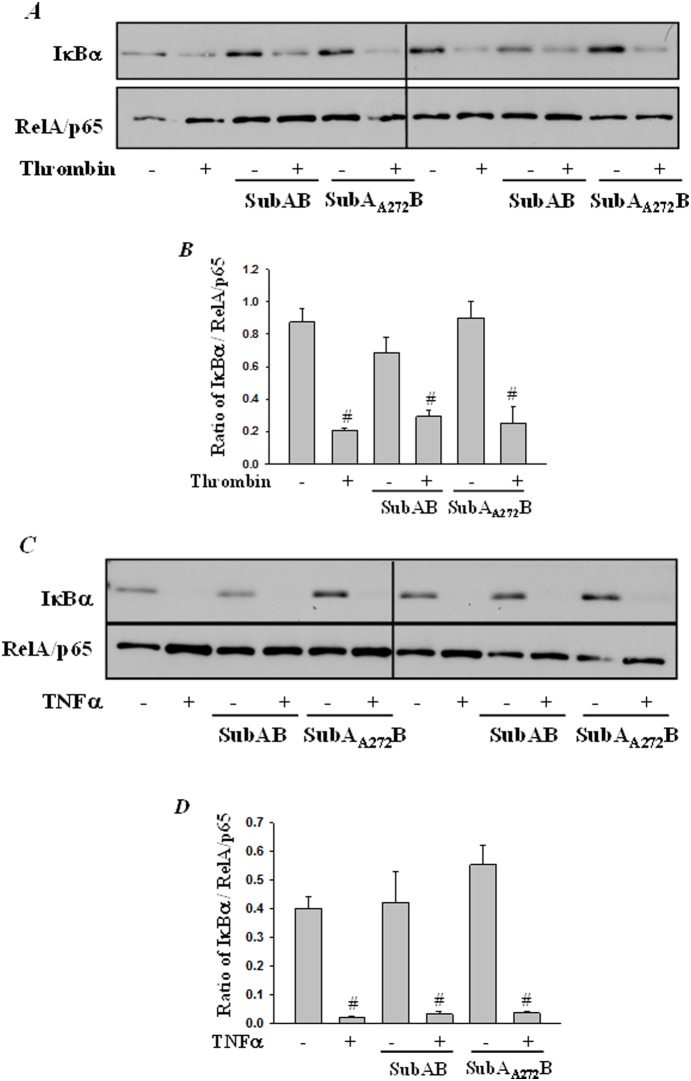
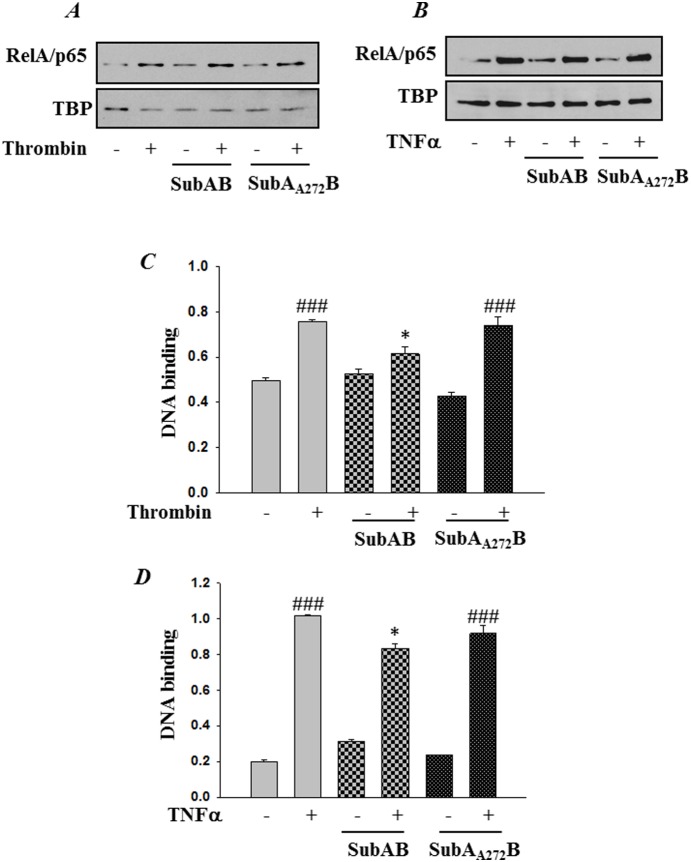
Endoplasmic Reticulum (ER) stress, caused by disturbance in ER homeostasis, has been implicated in several pathological conditions such as ischemic injury, neurodegenerative disorders, metabolic diseases and more recently in inflammatory conditions. Our present study aims at understanding the role of ER stress in endothelial cell (EC) inflammation, a critical event in the pathogenesis of acute lung injury (ALI). We found that preconditioning human pulmonary artery endothelial cells (HPAEC) to ER stress either by depleting ER chaperone and signaling regulator BiP using siRNA, or specifically cleaving (inactivating) BiP using subtilase cytotoxin (SubAB), alleviates EC inflammation. The two approaches adopted to abrogate BiP function induced ATF4 protein expression and the phosphorylation of eIF2α, both markers of ER stress, which in turn resulted in blunting the activation of NF-κB, and restoring endothelial barrier integrity. Pretreatment of HPAEC with BiP siRNA inhibited thrombin-induced IκBα degradation and its resulting downstream signaling pathway involving NF-κB nuclear translocation, DNA binding, phosphorylation at serine536, transcriptional activation and subsequent expression of adhesion molecules. However, TNFα-mediated NF-κB signaling was unaffected upon BiP knockdown. In an alternative approach, SubAB-mediated inactivation of NF-κB was independent of IκBα degradation. Mechanistic analysis revealed that pretreatment of EC with SubAB interfered with the binding of the liberated NF-κB to the DNA, thereby resulting in reduced expression of adhesion molecules, cytokines and chemokines. In addition, both knockdown and inactivation of BiP stimulated actin cytoskeletal reorganization resulting in restoration of endothelial permeability. Together our studies indicate that BiP plays a central role in EC inflammation and injury via its action on NF-κB activation and regulation of vascular permeability.
内质网(ER)稳态紊乱所导致的内质网应激与多种病理状况相关,如缺血性损伤、神经退行性疾病、代谢性疾病,以及最近发现的炎症状态。我们目前的研究旨在了解内质网应激在急性肺损伤(ALI)发病机制中的关键事件——内皮细胞(EC)炎症中的作用。我们发现,通过使用小干扰RNA(siRNA)消耗内质网伴侣和信号调节因子BiP,或使用枯草杆菌蛋白酶细胞毒素(SubAB)特异性切割(灭活)BiP,对人肺动脉内皮细胞(HPAEC)进行内质网应激预处理,可减轻内皮细胞炎症。采用的两种消除BiP功能的方法诱导了内质网应激的两个标志物——激活转录因子4(ATF4)蛋白表达和真核翻译起始因子2α(eIF2α)的磷酸化,这反过来又导致核因子κB(NF-κB)的激活减弱,并恢复内皮屏障完整性。用BiP siRNA预处理HPAEC可抑制凝血酶诱导的IκBα降解及其下游涉及NF-κB核转位、DNA结合、丝氨酸536磷酸化、转录激活以及随后粘附分子表达的信号通路。然而,BiP敲低后,肿瘤坏死因子α(TNFα)介导的NF-κB信号传导未受影响。在另一种方法中,SubAB介导的NF-κB失活与IκBα降解无关。机制分析表明,用SubAB预处理内皮细胞会干扰游离的NF-κB与DNA的结合,从而导致粘附分子、细胞因子和趋化因子的表达减少。此外,BiP的敲低和失活均刺激肌动蛋白细胞骨架重排,从而恢复内皮通透性。我们的研究共同表明,BiP通过其对NF-κB激活和血管通透性调节的作用,在内皮细胞炎症和损伤中起核心作用。