Gapsys Vytautas, Michielssens Servaas, Seeliger Daniel, de Groot Bert L
Computational Biomolecular Dynamics Group, Max Planck Institute for Biophysical Chemistry, Am Fassberg 11, Göttingen, 37077, Germany.
J Comput Chem. 2015 Feb 15;36(5):348-54. doi: 10.1002/jcc.23804. Epub 2014 Dec 8.
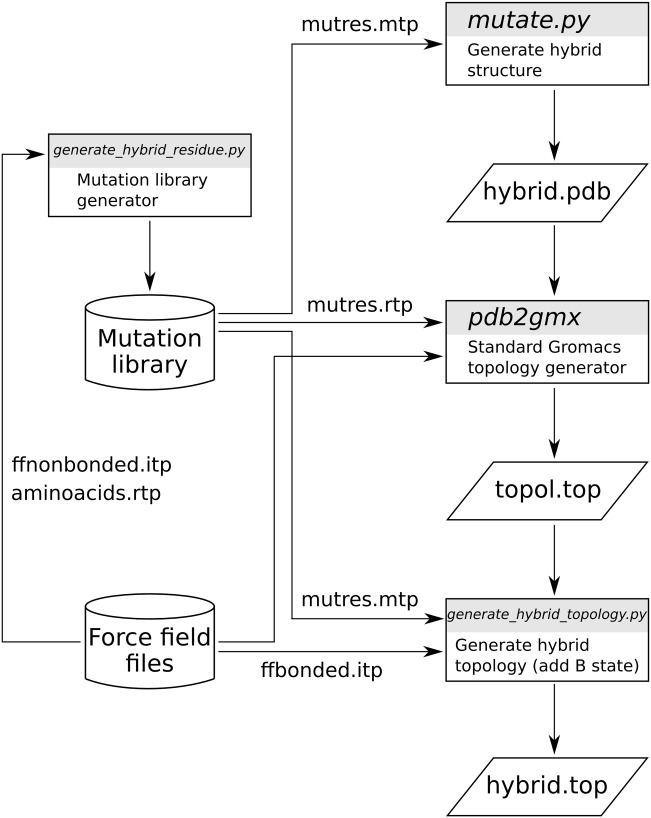
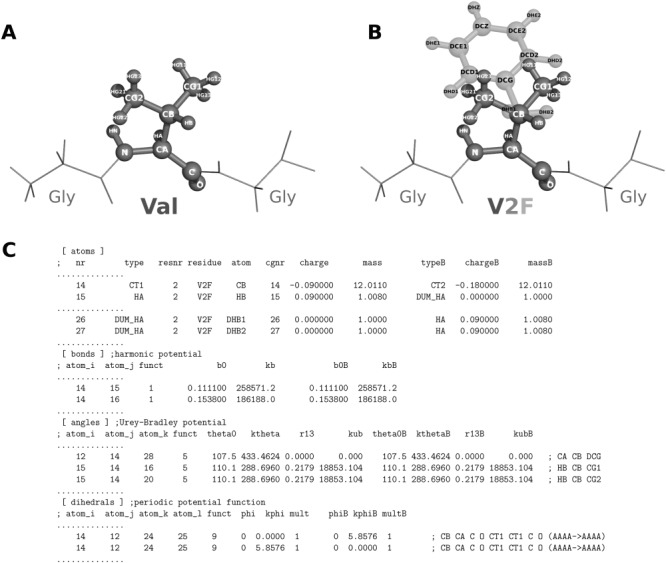
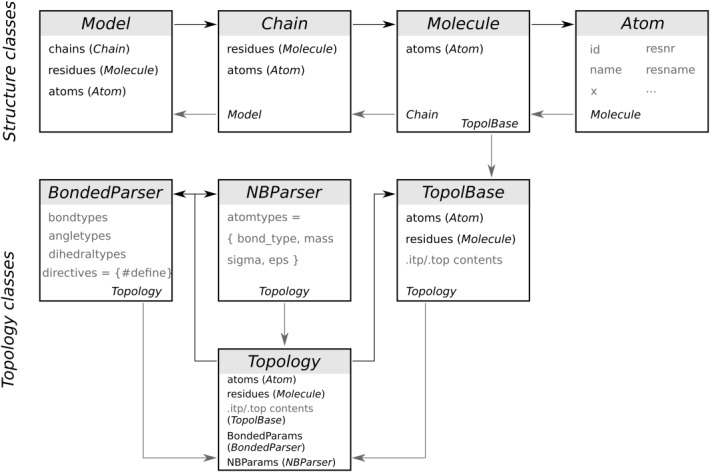
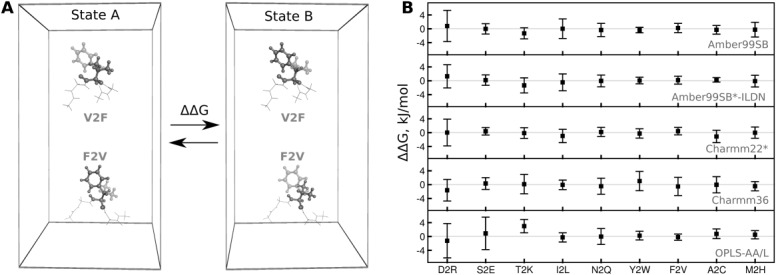
Computational protein design requires methods to accurately estimate free energy changes in protein stability or binding upon an amino acid mutation. From the different approaches available, molecular dynamics-based alchemical free energy calculations are unique in their accuracy and solid theoretical basis. The challenge in using these methods lies in the need to generate hybrid structures and topologies representing two physical states of a system. A custom made hybrid topology may prove useful for a particular mutation of interest, however, a high throughput mutation analysis calls for a more general approach. In this work, we present an automated procedure to generate hybrid structures and topologies for the amino acid mutations in all commonly used force fields. The described software is compatible with the Gromacs simulation package. The mutation libraries are readily supported for five force fields, namely Amber99SB, Amber99SB*-ILDN, OPLS-AA/L, Charmm22*, and Charmm36.
计算蛋白质设计需要能够准确估计氨基酸突变时蛋白质稳定性或结合自由能变化的方法。在现有的不同方法中,基于分子动力学的炼金术自由能计算在准确性和坚实的理论基础方面独具特色。使用这些方法的挑战在于需要生成代表系统两种物理状态的混合结构和拓扑结构。定制的混合拓扑结构可能对特定的感兴趣突变有用,然而,高通量突变分析需要一种更通用的方法。在这项工作中,我们提出了一种自动程序,用于为所有常用力场中的氨基酸突变生成混合结构和拓扑结构。所描述的软件与Gromacs模拟包兼容。该突变库可轻松支持五种力场,即Amber99SB、Amber99SB*-ILDN、OPLS-AA/L、Charmm22*和Charmm36。