Chou I-Ching, Lin Sheng-Shing, Lin Wei-De, Wang Chung-Hsing, Chang Yu-Tzu, Tsai Fuu-Jen, Tsai Chang-Hai
Department of Pediatrics, Children's Hospital, China Medical University Hospital, Taichung, Taiwan ; Graduate Institute of Integrated Medicine, College of Chinese Medicine, China Medical University, Taichung, Taiwan.
Department of Pediatrics, Children's Hospital, China Medical University Hospital, Taichung, Taiwan.
Biomedicine (Taipei). 2014;4(2):15. doi: 10.7603/s40681-014-0015-0. Epub 2014 May 8.
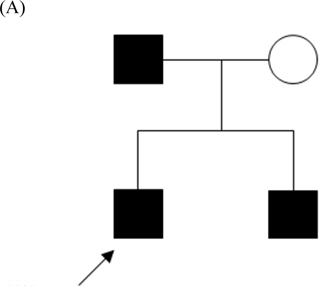
Paroxysmal kinesigenic dyskinesia (PKD), a rare paroxysmal movement disorder often misdiagnosed as epilepsy, is characterized by recurrent, brief dyskinesia attacks triggered by sudden voluntary movement. Pathophysiological mechanism of PKD remains not well understood. Ion channelopathy has been suggested, since the disease responds well to ion channel blockers. Mutations in proline-rich transmembrane protein 2 () were recently identified in patients with familial PKD. To extend these genetic reports, we studied a family with clinical manifestations of familial PKD responding well to low dose carbamazepine. Therapeutic dose ranged from 1.5 to 2.0 mg/ kg/day, below that in seizure control. One insertion mutation c.649_650insC (p.P217fsX7) was identified in three patients of the family. This study avers 's high sensitivity for PKD phenotype. Identification of genes underlying pathogenesis will enhance diagnosis and treatment. Function of and its role in PKD warrant further investigation.
发作性运动诱发性运动障碍(PKD)是一种罕见的发作性运动障碍,常被误诊为癫痫,其特征是由突然的自主运动引发反复发作的短暂运动障碍发作。PKD的病理生理机制仍未完全明确。由于该疾病对离子通道阻滞剂反应良好,因此有人提出离子通道病的观点。最近在家族性PKD患者中发现了富含脯氨酸的跨膜蛋白2()的突变。为了扩展这些遗传学报告,我们研究了一个具有家族性PKD临床表现且对低剂量卡马西平反应良好的家庭。治疗剂量为1.5至2.0毫克/千克/天,低于癫痫控制的剂量。在该家庭的三名患者中鉴定出一个插入突变c.649_650insC(p.P217fsX7)。本研究证实了对PKD表型具有高敏感性。确定发病机制的相关基因将有助于提高诊断和治疗水平。及其在PKD中的作用仍有待进一步研究。