Cui Di, Ou Shu-Ching, Patel Sandeep
Department of Chemistry and Biochemistry, University of Delaware , Newark, Delaware 19716, United States.
J Phys Chem B. 2015 Jan 8;119(1):164-78. doi: 10.1021/jp507203g. Epub 2014 Dec 23.
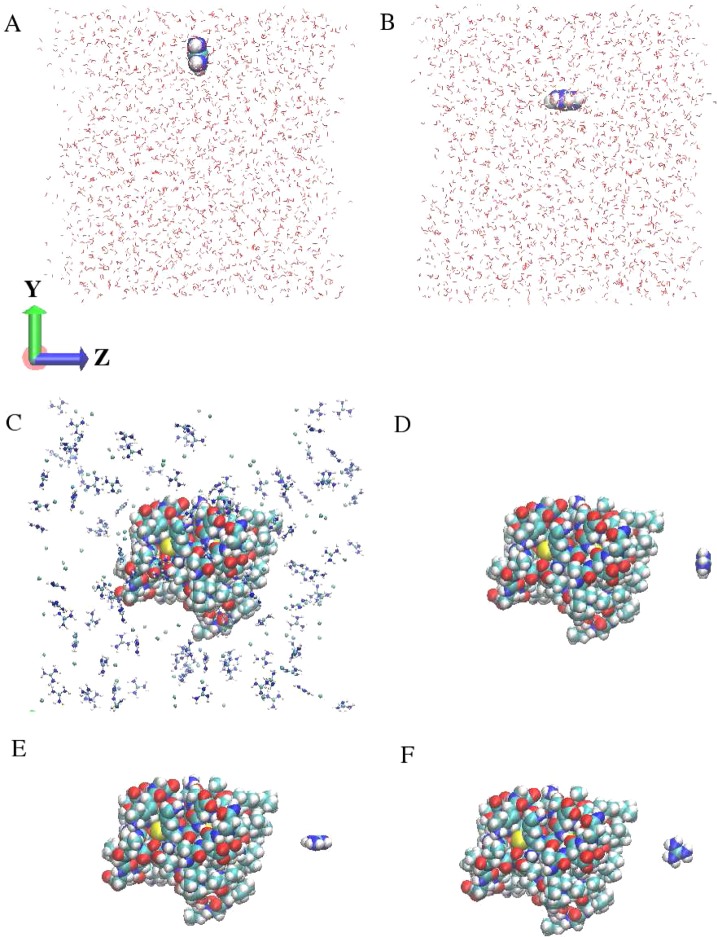
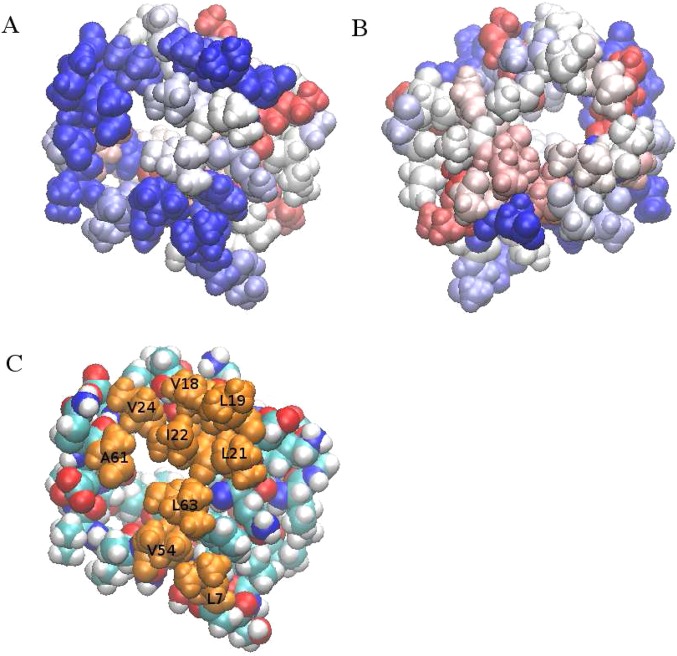
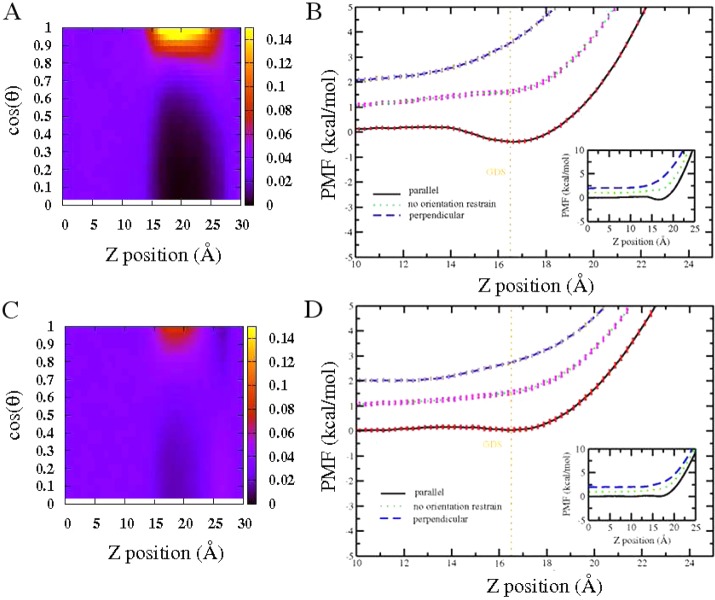
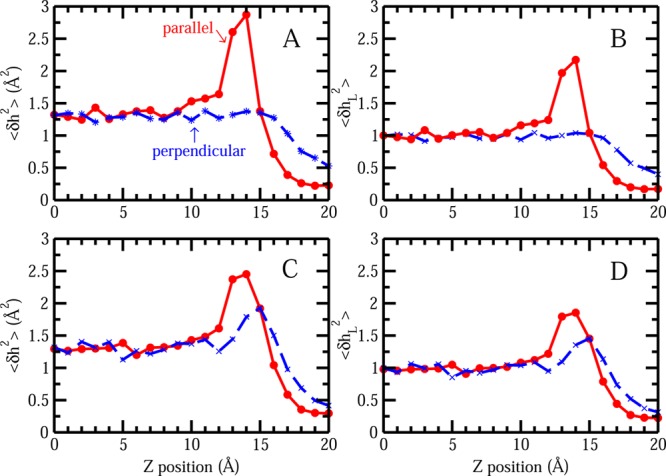
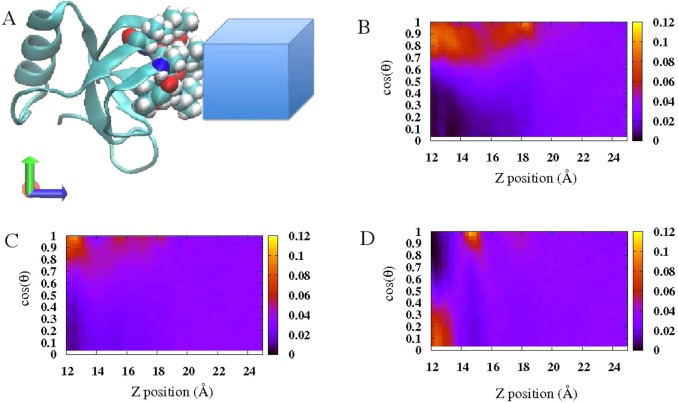
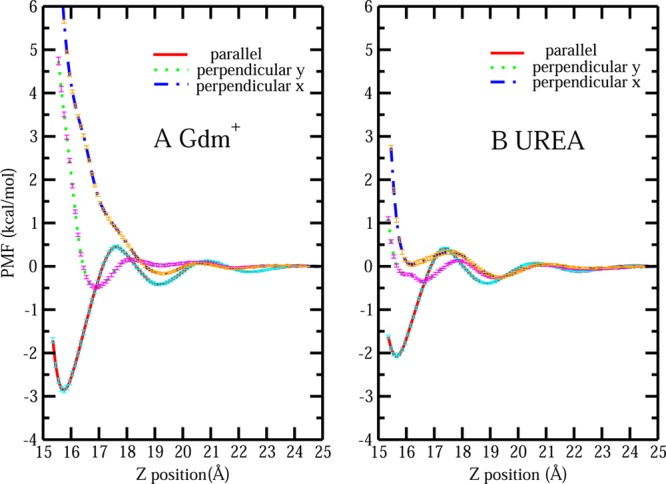
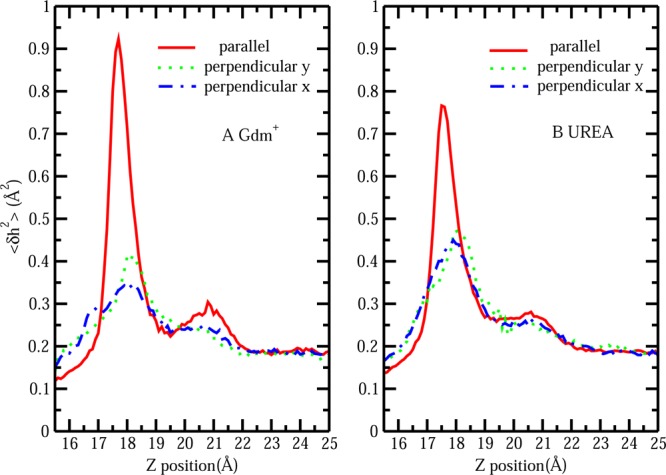
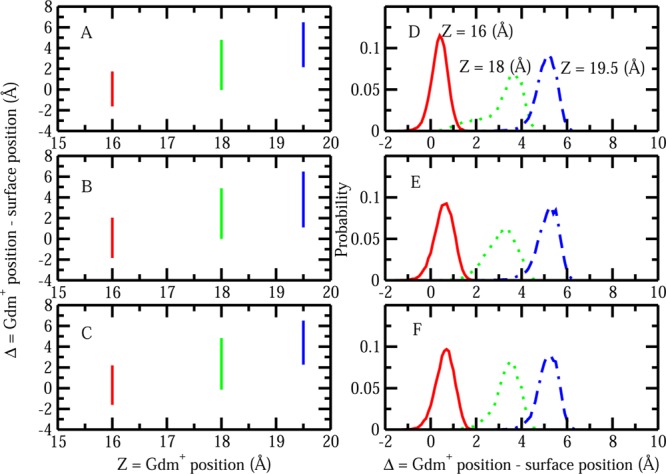
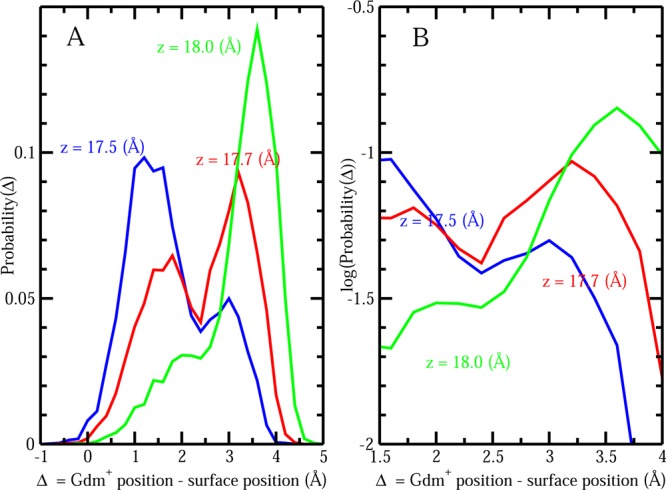
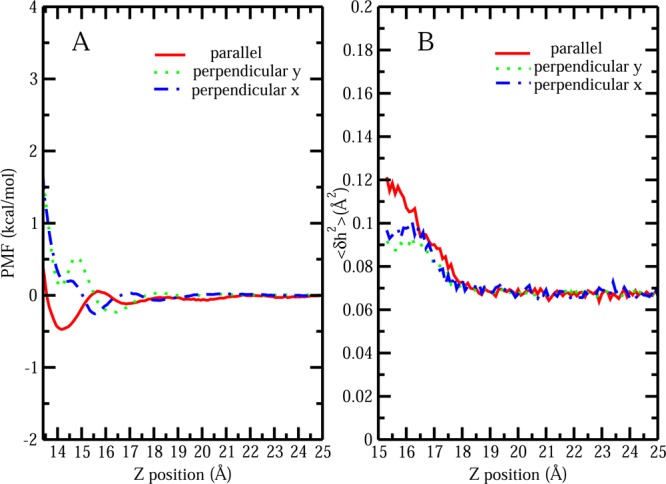
The notion of direct interaction between denaturing cosolvent and protein residues has been proposed in dialogue relevant to molecular mechanisms of protein denaturation. Here we consider the correlation between free energetic stability and induced fluctuations of an aqueous-hydrophobic interface between a model hydrophobically associating protein, HFBII, and two common protein denaturants, guanidinium cation (Gdm(+)) and urea. We compute potentials of mean force along an order parameter that brings the solute molecule close to the known hydrophobic region of the protein. We assess potentials of mean force for different relative orientations between the protein and denaturant molecule. We find that in both cases of guanidinium cation and urea relative orientations of the denaturant molecule that are parallel to the local protein-water interface exhibit greater stability compared to edge-on or perpendicular orientations. This behavior has been observed for guanidinium/methylguanidinium cations at the liquid-vapor interface of water, and thus the present results further corroborate earlier findings. Further analysis of the induced fluctuations of the aqueous-hydrophobic interface upon approach of the denaturant molecule indicates that the parallel orientation, displaying a greater stability at the interface, also induces larger fluctuations of the interface compared to the perpendicular orientations. The correlation of interfacial stability and induced interface fluctuation is a recurring theme for interface-stable solutes at hydrophobic interfaces. Moreover, observed correlations between interface stability and induced fluctuations recapitulate connections to local hydration structure and patterns around solutes as evidenced by experiment (Cooper et al., J. Phys. Chem. A 2014, 118, 5657.) and high-level ab initio/DFT calculations (Baer et al., Faraday Discuss 2013, 160, 89).
在与蛋白质变性分子机制相关的讨论中,有人提出变性助溶剂与蛋白质残基之间存在直接相互作用的观点。在此,我们考虑一种模型疏水缔合蛋白HFBII与两种常见蛋白质变性剂胍阳离子(Gdm(+))和尿素之间的水-疏水界面的自由能稳定性与诱导波动之间的相关性。我们沿着一个序参量计算平均力势,该序参量使溶质分子靠近蛋白质已知的疏水区域。我们评估蛋白质和变性剂分子之间不同相对取向的平均力势。我们发现,在胍阳离子和尿素这两种情况下,与边缘取向或垂直取向相比,变性剂分子与局部蛋白质-水界面平行的相对取向表现出更高的稳定性。在水的液-气界面上,胍/甲基胍阳离子也观察到了这种行为,因此目前的结果进一步证实了早期的发现。对变性剂分子靠近时水-疏水界面诱导波动的进一步分析表明,与垂直取向相比,在界面处表现出更高稳定性的平行取向也会诱导更大的界面波动。界面稳定性与诱导界面波动之间的相关性是疏水界面上界面稳定溶质的一个反复出现的主题。此外,观察到的界面稳定性与诱导波动之间的相关性概括了与溶质周围局部水合结构和模式的联系,实验(Cooper等人,《物理化学杂志A》2014年,118卷,5657页)和高水平的从头算/密度泛函理论计算(Baer等人,《法拉第讨论》2013年,160卷,89页)证明了这一点。