Endocrine Unit, University of California San Francisco and Veterans Affairs Medical Center San Francisco, California, 94121 ; Department of Neurology, University of California San Francisco and Veterans Affairs Medical Center San Francisco, California, 94121.
Endocrine Unit, University of California San Francisco and Veterans Affairs Medical Center San Francisco, California, 94121.
Ann Clin Transl Neurol. 2014 Nov;1(11):851-66. doi: 10.1002/acn3.118. Epub 2014 Oct 3.
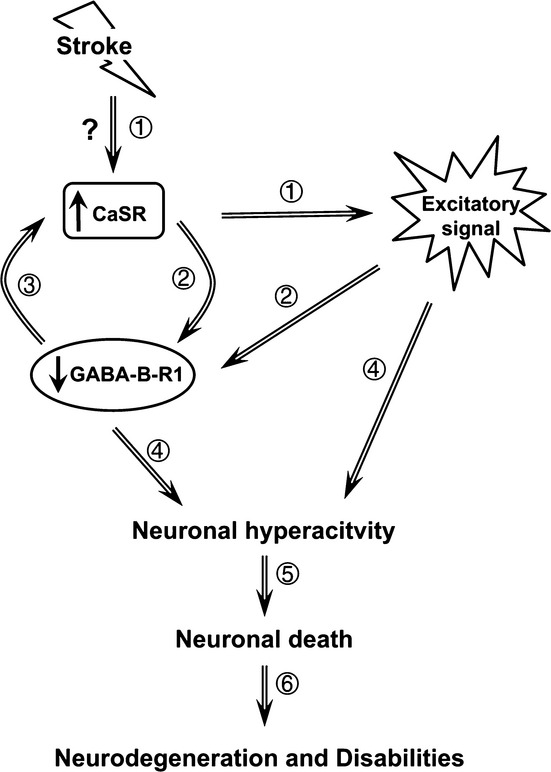
Ischemic brain injury is the leading cause for death and long-term disability in patients who suffer cardiac arrest and embolic stroke. Excitotoxicity and subsequent Ca(2+)-overload lead to ischemic neuron death. We explore a novel mechanism concerning the role of the excitatory extracellular calcium-sensing receptor (CaSR) in the induction of ischemic brain injury.
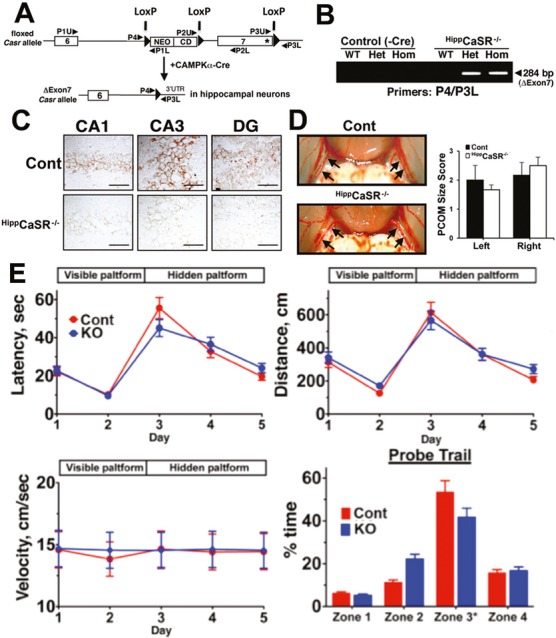
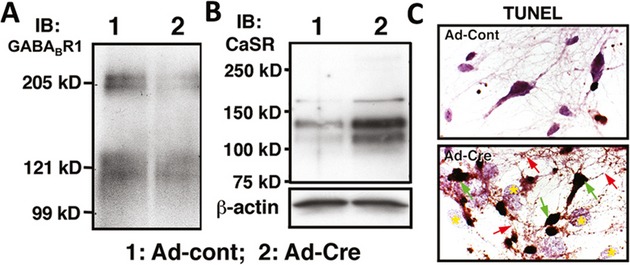
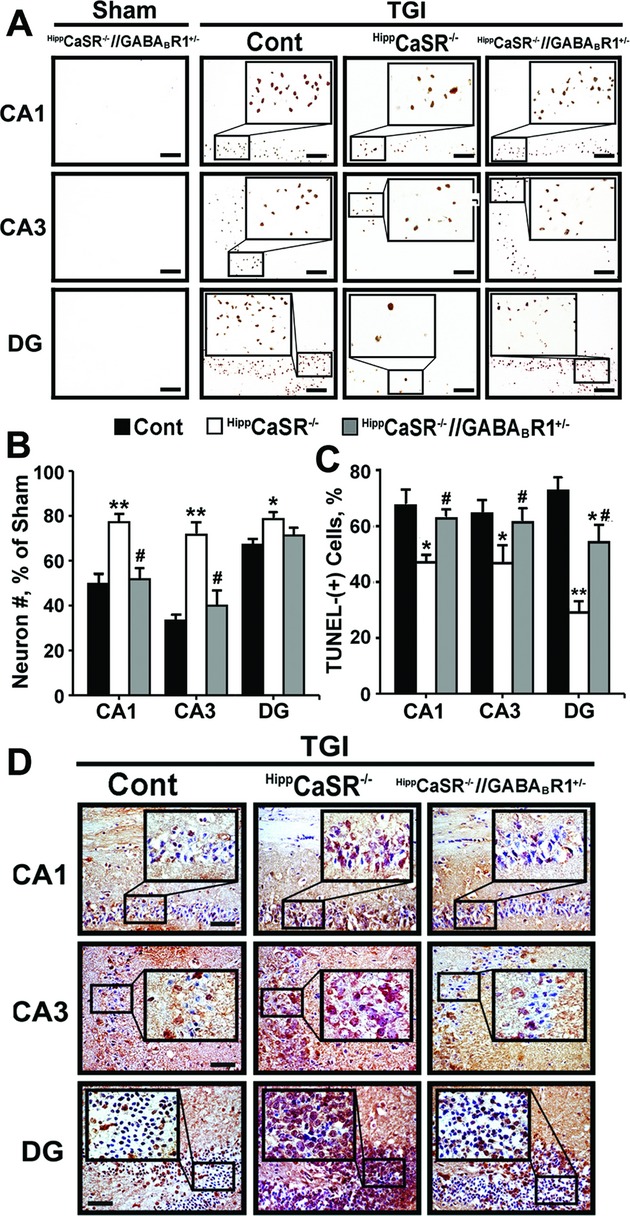
Mice were exposed to forebrain ischemia and the actions of CaSR were determined after its genes were ablated specifically in hippocampal neurons or its activities were inhibited pharmacologically. Since the CaSR forms a heteromeric complex with the inhibitory type B γ-aminobutyric acid receptor 1 (GABABR1), we compared neuronal responses to ischemia in mice deficient in CaSR, GABABR1, or both, and in mice injected locally or systemically with a specific CaSR antagonist (or calcilytic) in the presence or absence of a GABABR1 agonist (baclofen).
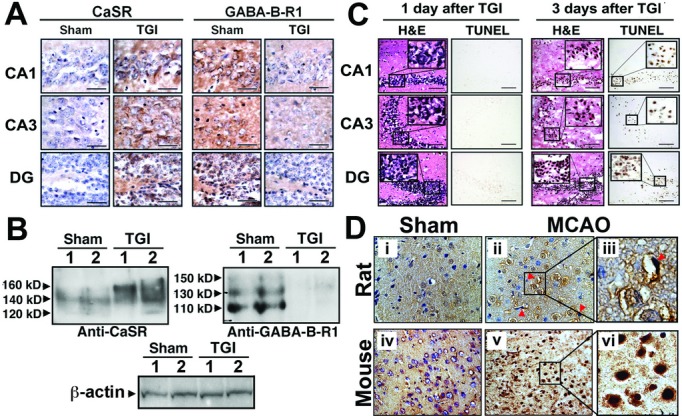
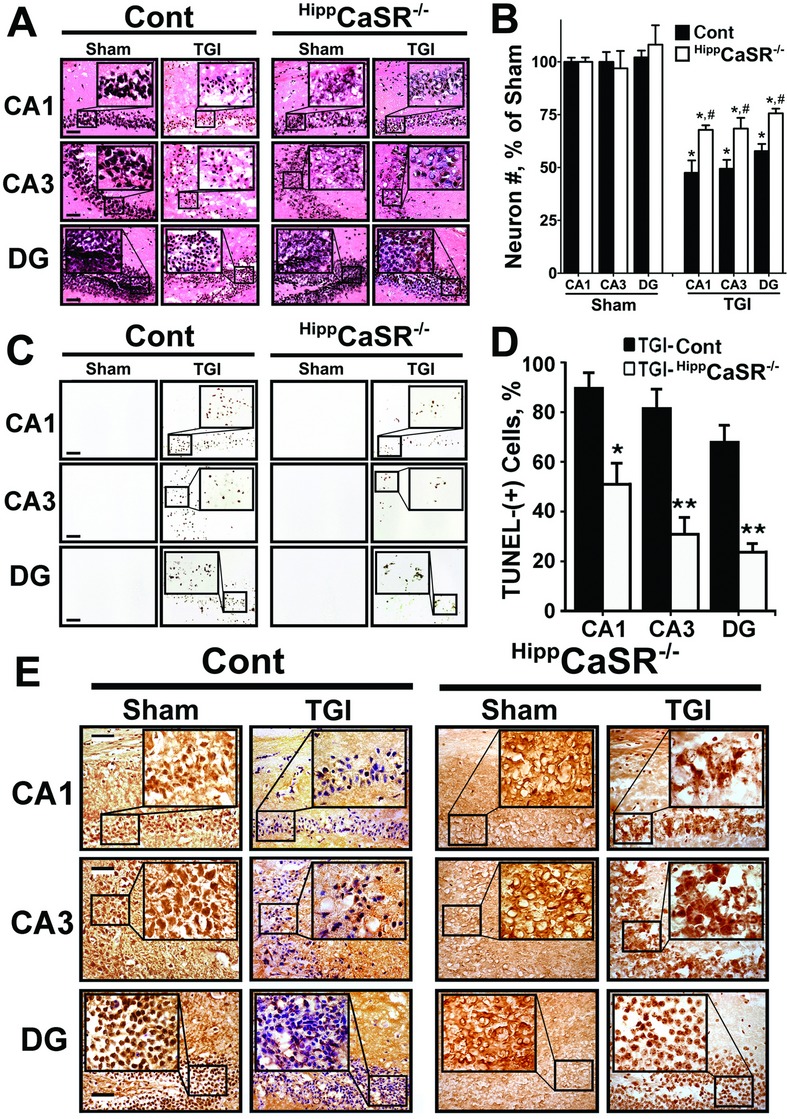
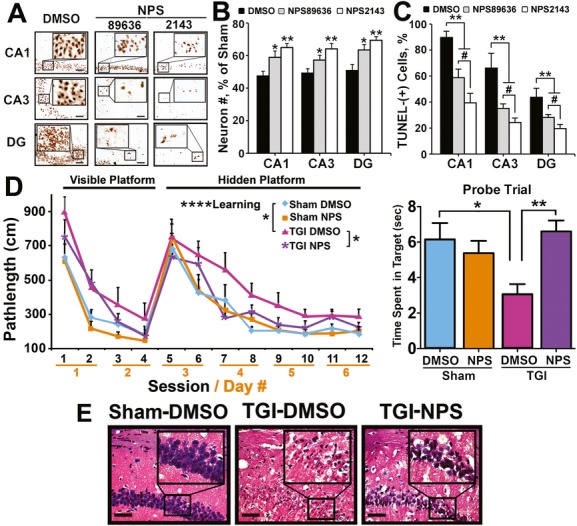
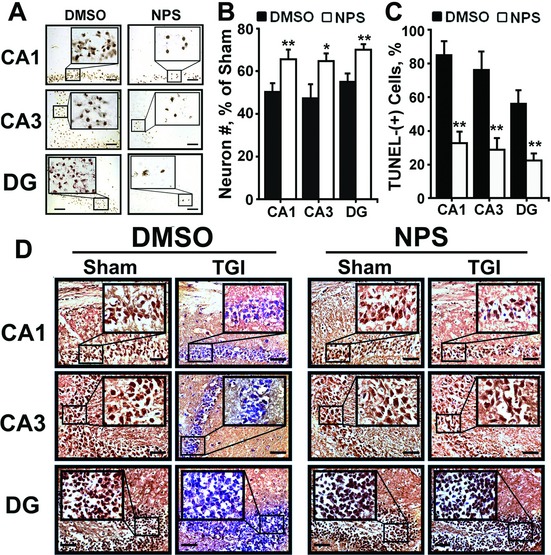
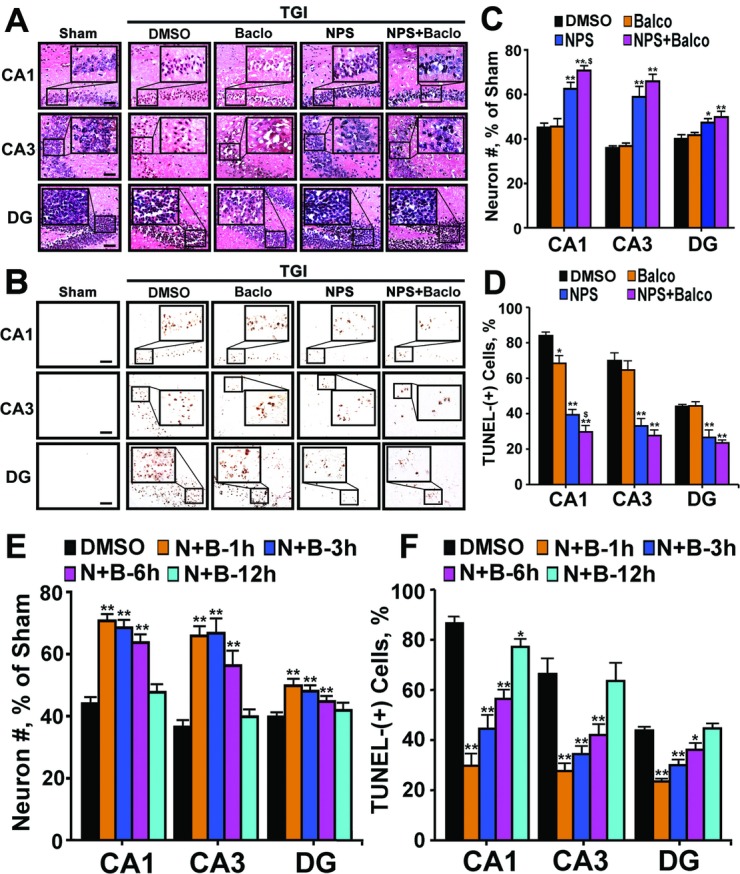
Both global and focal brain ischemia led to CaSR overexpression and GABABR1 downregulation in injured neurons. Genetic ablation of Casr genes or blocking CaSR activities by calcilytics rendered robust neuroprotection and preserved learning and memory functions in ischemic mice, partly by restoring GABABR1 expression. Concurrent ablation of Gabbr1 gene blocked the neuroprotection caused by the Casr gene knockout. Coinjection of calcilytics with baclofen synergistically enhanced neuroprotection. This combined therapy remained robust when given 6 h after ischemia.
Our study demonstrates a novel receptor interaction, which contributes to ischemic neuron death through CaSR upregulation and GABABR1 downregulation, and feasibility of neuroprotection by concurrently targeting these two receptors.
缺血性脑损伤是导致心脏骤停和栓塞性中风患者死亡和长期残疾的主要原因。兴奋性毒性和随后的 Ca(2+)-过载导致缺血性神经元死亡。我们探索了一种关于兴奋性细胞外钙敏感受体 (CaSR) 在诱导缺血性脑损伤中的作用的新机制。
将小鼠暴露于前脑缺血,并在其基因特异性在海马神经元中缺失或其活性被药理学抑制后确定 CaSR 的作用。由于 CaSR 与抑制型 B 型 γ-氨基丁酸受体 1 (GABABR1) 形成异源三聚体复合物,我们比较了 CaSR 缺失、GABABR1 缺失或两者均缺失的小鼠以及局部或系统注射特定 CaSR 拮抗剂(或钙敏剂)的小鼠在存在或不存在 GABABR1 激动剂 (baclofen) 时对缺血的神经元反应。
全脑和局灶性脑缺血导致损伤神经元中 CaSR 过表达和 GABABR1 下调。Casr 基因缺失或钙敏剂阻断 CaSR 活性使缺血小鼠具有强大的神经保护作用,并保留学习和记忆功能,部分是通过恢复 GABABR1 表达。同时缺失 Gabbr1 基因阻断了 Casr 基因敲除引起的神经保护作用。钙敏剂与 baclofen 联合注射协同增强神经保护作用。这种联合治疗在缺血后 6 小时给予时仍然有效。
我们的研究表明了一种新的受体相互作用,通过 CaSR 上调和 GABABR1 下调导致缺血性神经元死亡,并通过同时针对这两个受体进行神经保护的可行性。