Department of Computer Science and Engineering, University of Minnesota, Minneapolis, Minnesota 55455 USA ; Broad Institute of Harvard and MIT, Cambridge, Massachusetts 02142 USA ; Center for Computational and Integrative Biology, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts 02114 USA ; Biotechnology Institute, University of Minnesota, St. Paul, Minnesota 55108 USA.
Zane Cohen Centre for Digestive Diseases, Mount Sinai Hospital IBD Group, University of Toronto, Toronto, Ontario M5G 1X5 Canada.
Genome Med. 2014 Dec 2;6(12):107. doi: 10.1186/s13073-014-0107-1. eCollection 2014.
Human genetics and host-associated microbial communities have been associated independently with a wide range of chronic diseases. One of the strongest associations in each case is inflammatory bowel disease (IBD), but disease risk cannot be explained fully by either factor individually. Recent findings point to interactions between host genetics and microbial exposures as important contributors to disease risk in IBD. These include evidence of the partial heritability of the gut microbiota and the conferral of gut mucosal inflammation by microbiome transplant even when the dysbiosis was initially genetically derived. Although there have been several tests for association of individual genetic loci with bacterial taxa, there has been no direct comparison of complex genome-microbiome associations in large cohorts of patients with an immunity-related disease.
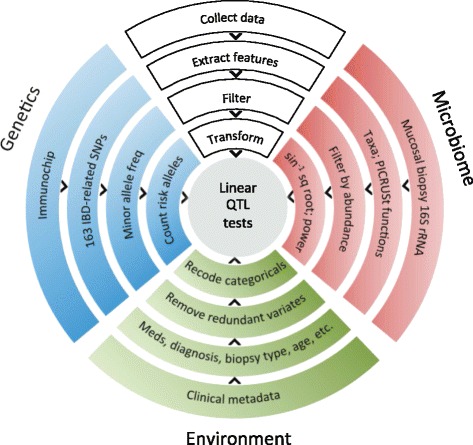
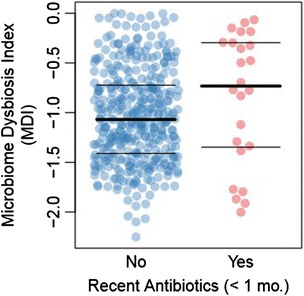
We obtained 16S ribosomal RNA (rRNA) gene sequences from intestinal biopsies as well as host genotype via Immunochip in three independent cohorts totaling 474 individuals. We tested for correlation between relative abundance of bacterial taxa and number of minor alleles at known IBD risk loci, including fine mapping of multiple risk alleles in the Nucleotide-binding oligomerization domain-containing protein 2 (NOD2) gene exon. We identified host polymorphisms whose associations with bacterial taxa were conserved across two or more cohorts, and we tested related genes for enrichment of host functional pathways.
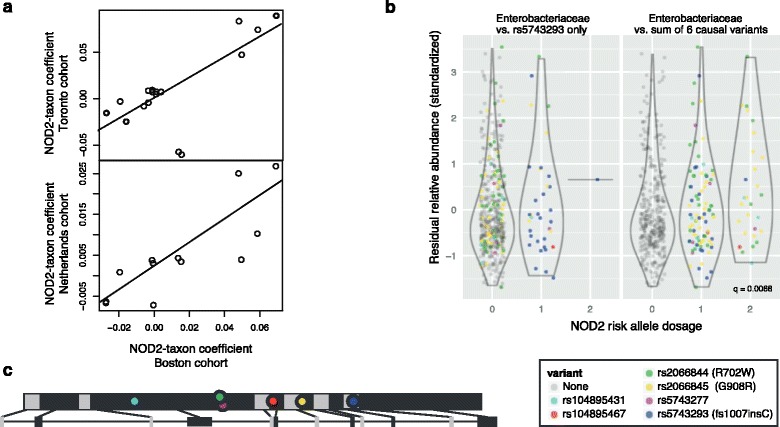
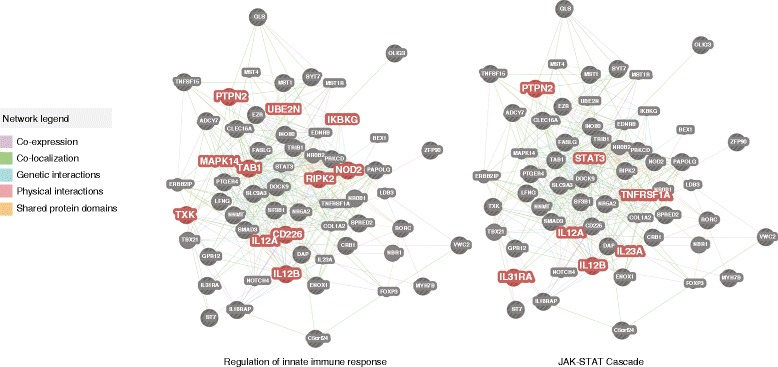
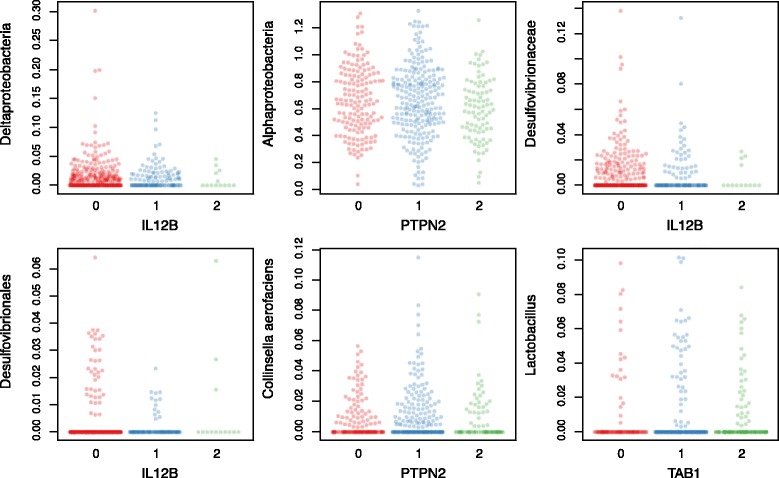
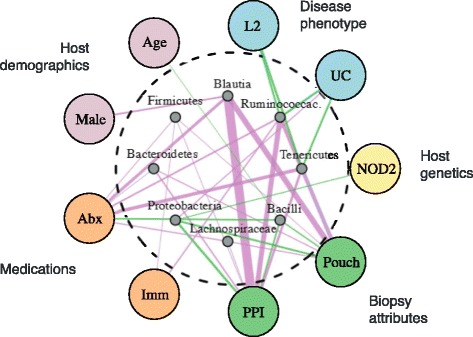
We identified and confirmed in two cohorts a significant association between NOD2 risk allele count and increased relative abundance of Enterobacteriaceae, with directionality of the effect conserved in the third cohort. Forty-eight additional IBD-related SNPs have directionality of their associations with bacterial taxa significantly conserved across two or three cohorts, implicating genes enriched for regulation of innate immune response, the JAK-STAT cascade, and other immunity-related pathways.
These results suggest complex interactions between genetically altered host functional pathways and the structure of the microbiome. Our findings demonstrate the ability to uncover novel associations from paired genome-microbiome data, and they suggest a complex link between host genetics and microbial dysbiosis in subjects with IBD across independent cohorts.
人类遗传学和宿主相关的微生物群落与广泛的慢性疾病独立相关。在每种情况下,最强的关联之一是炎症性肠病(IBD),但仅凭这两个因素都无法完全解释疾病风险。最近的研究结果表明,宿主遗传学和微生物暴露之间的相互作用是 IBD 疾病风险的重要因素。这些相互作用包括肠道微生物组部分遗传可变性的证据,以及即使最初的微生物失调是遗传来源的,微生物组移植也能引发肠道粘膜炎症。尽管已经有几项针对个体遗传位点与细菌分类群关联的测试,但在具有免疫相关疾病的大患者队列中,还没有直接比较复杂的基因组-微生物组关联。
我们从三个独立的队列中的 474 名个体中通过免疫芯片获得了肠道活检的 16S 核糖体 RNA(rRNA)基因序列和宿主基因型。我们测试了细菌分类群的相对丰度与已知 IBD 风险基因座的少数等位基因数量之间的相关性,包括核苷酸结合寡聚结构域蛋白 2(NOD2)基因外显子中多个风险等位基因的精细定位。我们确定了在两个或更多队列中与细菌分类群相关的宿主多态性具有保守性,并测试了相关基因对宿主功能途径的富集情况。
我们在两个队列中鉴定并证实了 NOD2 风险等位基因计数与肠杆菌科相对丰度增加之间的显著关联,这种效应的方向性在第三个队列中保持一致。另外 48 个 IBD 相关 SNP 与细菌分类群的关联方向在两个或三个队列中具有显著的保守性,表明与先天免疫反应、JAK-STAT 级联和其他免疫相关途径的调节相关的基因富集。
这些结果表明,遗传改变的宿主功能途径与微生物组的结构之间存在复杂的相互作用。我们的研究结果表明,从配对的基因组-微生物组数据中可以发现新的关联,并表明在具有 IBD 的独立队列中,宿主遗传学和微生物失调之间存在复杂的联系。