Moore Z, Chakrabarti G, Luo X, Ali A, Hu Z, Fattah F J, Vemireddy R, DeBerardinis R J, Brekken R A, Boothman D A
Pharmacology and Radiation Oncology, Simmons Comprehensive Cancer Center, University of Texas Southwestern Medical Center, Dallas, TX, USA.
Internal Medicine and Touchstone Diabetes Center, Simmons Comprehensive Cancer Center, University of Texas Southwestern Medical Center, Dallas, TX, USA.
Cell Death Dis. 2015 Jan 15;6(1):e1599. doi: 10.1038/cddis.2014.564.
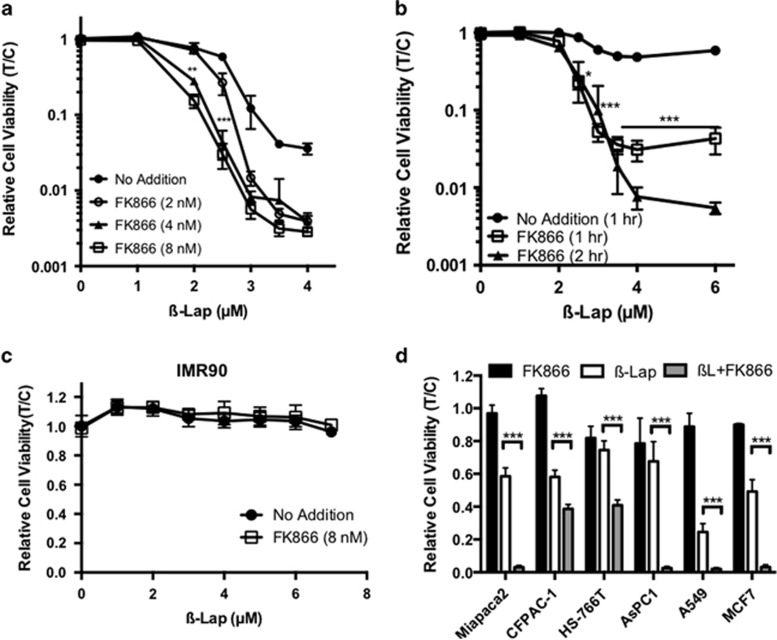
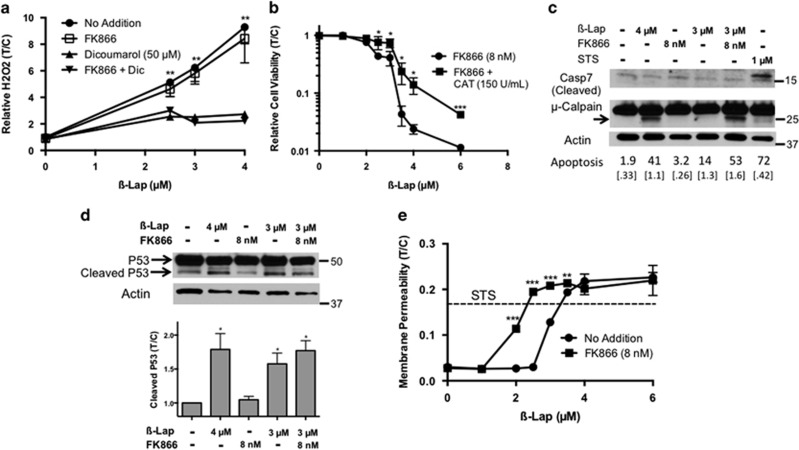
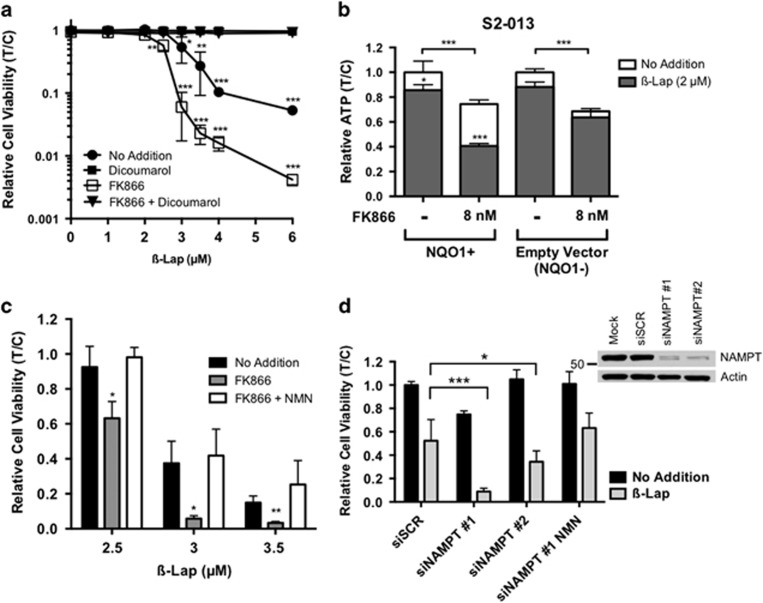
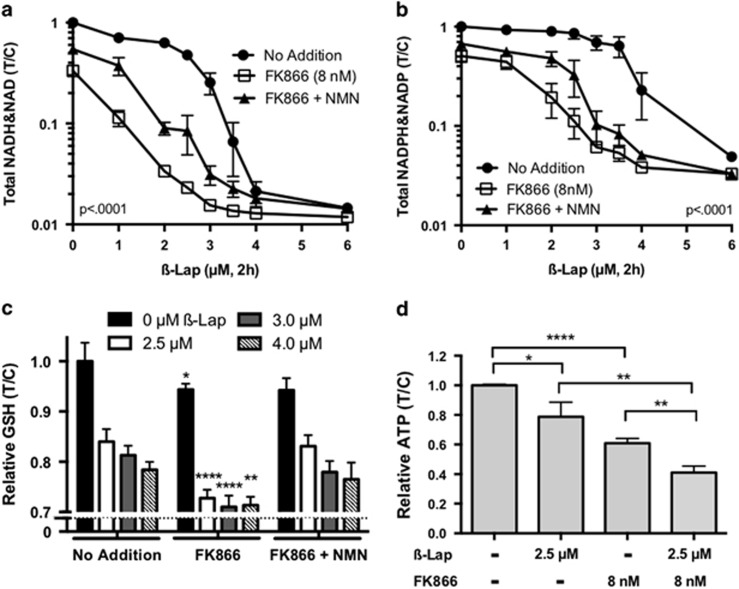
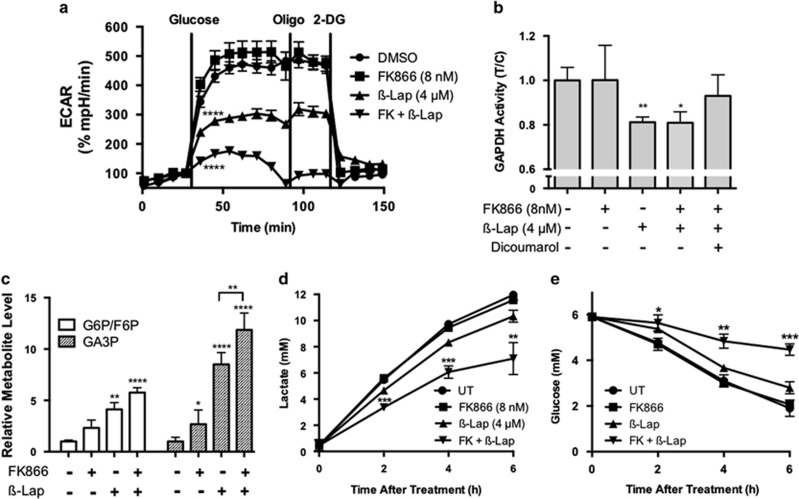
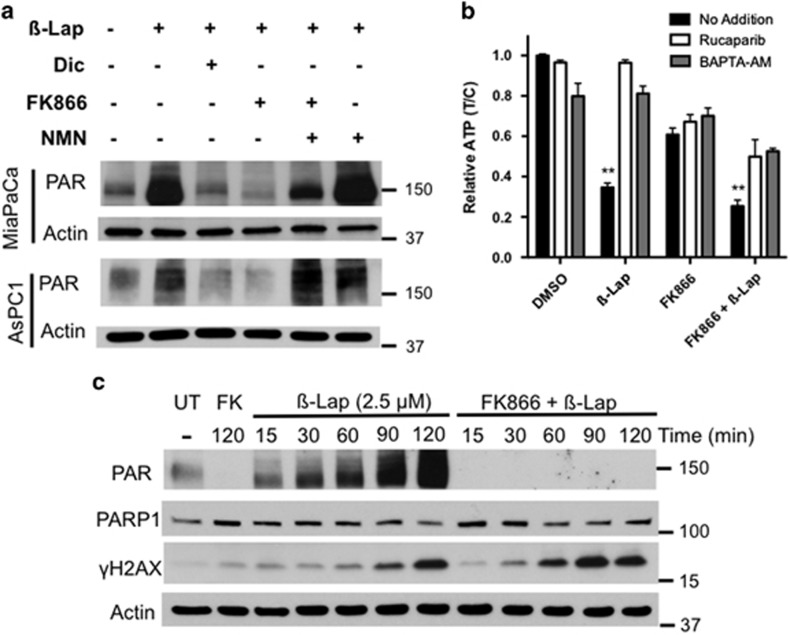
Nicotinamide phosphoribosyltransferase (NAMPT) inhibitors (e.g., FK866) target the most active pathway of NAD(+) synthesis in tumor cells, but lack tumor-selectivity for use as a single agent. Reducing NAD(+) pools by inhibiting NAMPT primed pancreatic ductal adenocarcinoma (PDA) cells for poly(ADP ribose) polymerase (PARP1)-dependent cell death induced by the targeted cancer therapeutic, β-lapachone (β-lap, ARQ761), independent of poly(ADP ribose) (PAR) accumulation. β-Lap is bioactivated by NADPH:quinone oxidoreductase 1 (NQO1) in a futile redox cycle that consumes oxygen and generates high levels of reactive oxygen species (ROS) that cause extensive DNA damage and rapid PARP1-mediated NAD(+) consumption. Synergy with FK866+β-lap was tumor-selective, only occurring in NQO1-overexpressing cancer cells, which is noted in a majority (∼85%) of PDA cases. This treatment strategy simultaneously decreases NAD(+) synthesis while increasing NAD(+) consumption, reducing required doses and treatment times for both drugs and increasing potency. These complementary mechanisms caused profound NAD(P)(+) depletion and inhibited glycolysis, driving down adenosine triphosphate levels and preventing recovery normally observed with either agent alone. Cancer cells died through an ROS-induced, μ-calpain-mediated programmed cell death process that kills independent of caspase activation and is not driven by PAR accumulation, which we call NAD(+)-Keresis. Non-overlapping specificities of FK866 for PDA tumors that rely heavily on NAMPT-catalyzed NAD(+) synthesis and β-lap for cancer cells with elevated NQO1 levels affords high tumor-selectivity. The concept of reducing NAD(+) pools in cancer cells to sensitize them to ROS-mediated cell death by β-lap is a novel strategy with potential application for pancreatic and other types of NQO1+ solid tumors.
烟酰胺磷酸核糖转移酶(NAMPT)抑制剂(如FK866)靶向肿瘤细胞中NAD(+)合成的最活跃途径,但作为单一药物缺乏肿瘤选择性。通过抑制NAMPT来降低NAD(+)水平,可使胰腺导管腺癌(PDA)细胞对靶向癌症治疗药物β-拉帕醌(β-lap,ARQ761)诱导的聚(ADP核糖)聚合酶(PARP1)依赖性细胞死亡敏感,且不依赖于聚(ADP核糖)(PAR)的积累。β-拉帕醌在一个无效的氧化还原循环中被NADPH:醌氧化还原酶1(NQO1)生物激活,该循环消耗氧气并产生高水平的活性氧(ROS),导致广泛的DNA损伤和PARP1介导的NAD(+)快速消耗。FK866与β-拉帕醌的协同作用具有肿瘤选择性,仅在NQO1过表达的癌细胞中出现,大多数(约85%)的PDA病例中都有这种情况。这种治疗策略同时降低了NAD(+)合成,同时增加了NAD(+)消耗,减少了两种药物所需的剂量和治疗时间,并提高了效力。这些互补机制导致了深刻的NAD(P)(+)耗竭并抑制了糖酵解,降低了三磷酸腺苷水平,并阻止了单独使用任何一种药物时通常观察到的恢复。癌细胞通过ROS诱导的、μ-钙蛋白酶介导的程序性细胞死亡过程死亡,该过程不依赖于半胱天冬酶激活,也不是由PAR积累驱动的,我们将其称为NAD(+)-Keresis。FK866对严重依赖NAMPT催化的NAD(+)合成的PDA肿瘤具有非重叠特异性,而β-拉帕醌对NQO1水平升高的癌细胞具有特异性,从而提供了高肿瘤选择性。降低癌细胞中NAD(+)水平以使其对β-拉帕醌介导的ROS介导的细胞死亡敏感的概念是一种新策略,具有应用于胰腺和其他类型NQO1+实体瘤的潜力。