Lin Runmao, Liu Chichuan, Shen Baoming, Bai Miao, Ling Jian, Chen Guohua, Mao Zhenchuan, Cheng Xinyue, Xie Bingyan
Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing, 100081, China.
College of Plant Protection, Hunan Agricultural University, Changsha, Hunan Province, 410128, China.
BMC Microbiol. 2015 Jan 31;15:5. doi: 10.1186/s12866-015-0341-8.
The fungus Pochonia chlamydosporia parasitizes nematode eggs and has become one of the most promising biological control agents (BCAs) for plant-parasitic nematodes, which are major agricultural pests that cause tremendous economic losses worldwide. The complete mitochondrial (mt) genome is expected to open new avenues for understanding the phylogenetic relationships and evolution of the invertebrate-pathogenic fungi in Hypocreales.
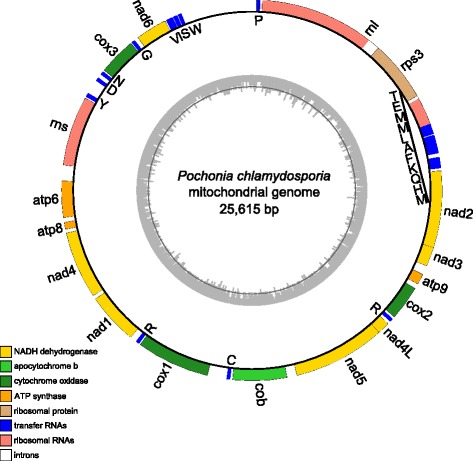
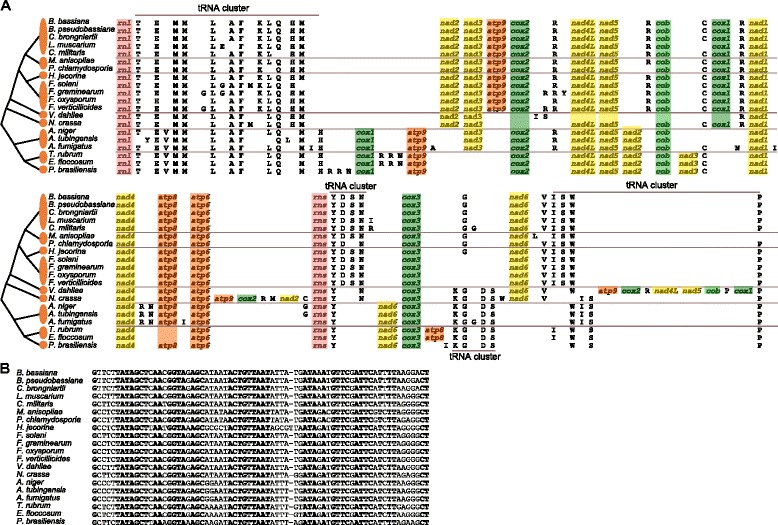
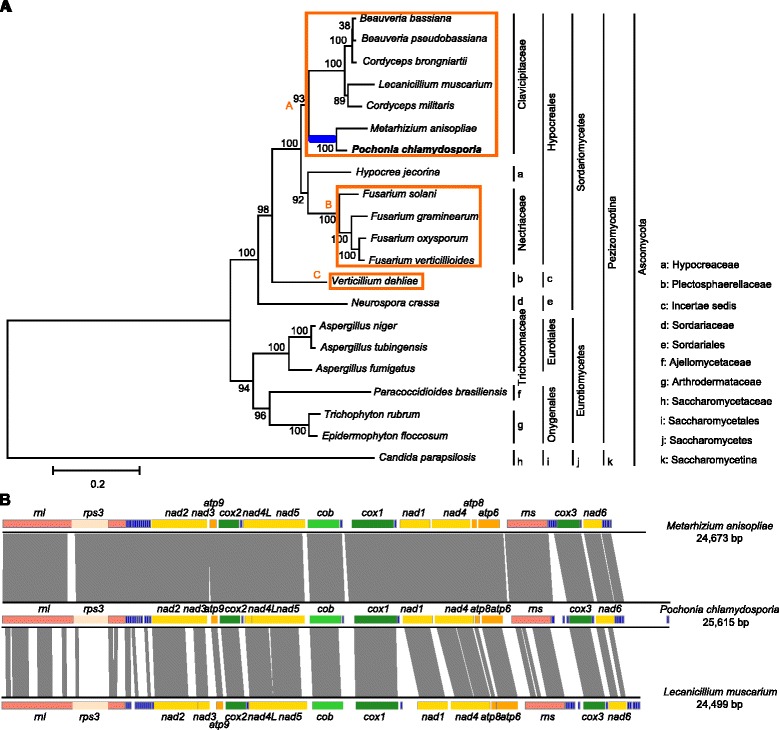
The complete mitogenome sequence of P. chlamydosporia is 25,615 bp in size, containing the 14 typical protein-coding genes, two ribosomal RNA genes, an intronic ORF coding for a putative ribosomal protein (rps3) and a set of 23 transfer RNA genes (trn) which recognize codons for all amino acids. Sequence similarity studies and syntenic gene analyses show that 87.02% and 58.72% of P. chlamydosporia mitogenome sequences match 90.50% of Metarhizium anisopliae sequences and 61.33% of Lecanicillium muscarium sequences with 92.38% and 86.04% identities, respectively. A phylogenetic tree inferred from 14 mt proteins in Pezizomycotina fungi supports that P. chlamydosporia is most closely related to the entomopathogenic fungus M. anisopliae. The invertebrate-pathogenic fungi in Hypocreales cluster together and clearly separate from a cluster comprising plant-pathogenic fungi (Fusarium spp.) and Hypocrea jecorina. A comparison of mitogenome sizes shows that the length of the intergenic regions or the intronic regions is the major size contributor in most of mitogenomes in Sordariomycetes. Evolutionary analysis shows that rps3 is under positive selection, leading to the display of unique evolutionary characteristics in Hypocreales. Moreover, the variability of trn distribution has a clear impact on gene order in mitogenomes. Gene rearrangement analysis shows that operation of transposition drives the rearrangement events in Pezizomycotina, and most events involve in trn position changes, but no rearrangement was found in Clavicipitaceae.
We present the complete annotated mitogenome sequence of P. chlamydosporia. Based on evolutionary and phylogenetic analyses, we have determined the relationships between the invertebrate-pathogenic fungi in Hypocreales. The invertebrate-pathogenic fungi in Hypocreales referred to in this paper form a monophyletic group sharing a most recent common ancestor. Our rps3 and trn gene order results also establish a foundation for further exploration of the evolutionary trajectory of the fungi in Hypocreales.
厚垣普可尼亚菌可寄生于线虫卵,已成为最具潜力的植物寄生线虫生物防治剂之一,植物寄生线虫是造成全球巨大经济损失的主要农业害虫。完整的线粒体(mt)基因组有望为理解肉座菌目无脊椎动物致病真菌的系统发育关系和进化开辟新途径。
厚垣普可尼亚菌的完整线粒体基因组序列大小为25,615 bp,包含14个典型的蛋白质编码基因、两个核糖体RNA基因、一个编码假定核糖体蛋白(rps3)的内含子开放阅读框以及一组23个转运RNA基因(trn),这些基因可识别所有氨基酸的密码子。序列相似性研究和共线性基因分析表明,厚垣普可尼亚菌线粒体基因组序列的87.02%和58.72%分别与绿僵菌序列的90.50%和蜡蚧轮枝菌序列的61.33%匹配,同一性分别为92.38%和86.04%。基于盘菌亚门真菌中14种线粒体蛋白推断的系统发育树支持厚垣普可尼亚菌与昆虫病原真菌绿僵菌关系最为密切。肉座菌目的无脊椎动物致病真菌聚在一起,明显与包括植物致病真菌(镰刀菌属)和嗜热毁丝霉在内的一个类群分开。线粒体基因组大小比较表明,基因间隔区或内含子区的长度是粪壳菌纲大多数线粒体基因组大小的主要贡献因素。进化分析表明,rps3受到正选择,导致肉座菌目呈现独特的进化特征。此外,trn分布的变异性对线粒体基因组中的基因顺序有明显影响。基因重排分析表明,转座作用驱动了盘菌亚门中的重排事件,大多数事件涉及trn位置变化,但在麦角菌科中未发现重排。
我们公布了厚垣普可尼亚菌完整的注释线粒体基因组序列。基于进化和系统发育分析,我们确定了肉座菌目无脊椎动物致病真菌之间的关系。本文所指的肉座菌目无脊椎动物致病真菌形成一个单系类群,共享一个最近的共同祖先。我们的rps3和trn基因顺序结果也为进一步探索肉座菌目真菌的进化轨迹奠定了基础。