Delaneau Olivier, Marchini Jonathan
Department of Statistics, University of Oxford, Oxford OX1 3TG, UK.
1] Department of Statistics, University of Oxford, Oxford OX1 3TG, UK [2] Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford OX3 7BN, UK.
Nat Commun. 2014 Jun 13;5:3934. doi: 10.1038/ncomms4934.
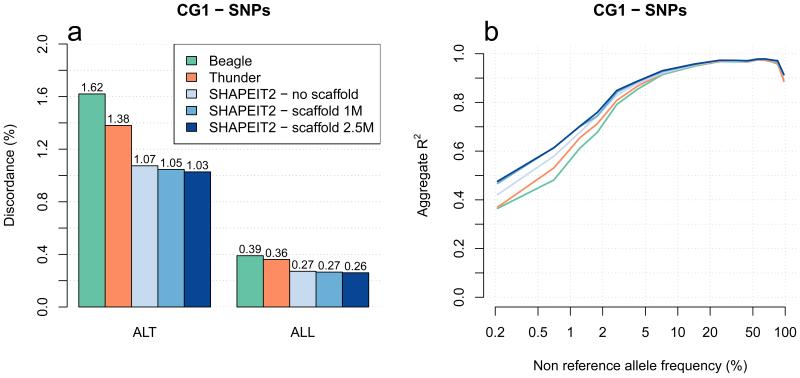
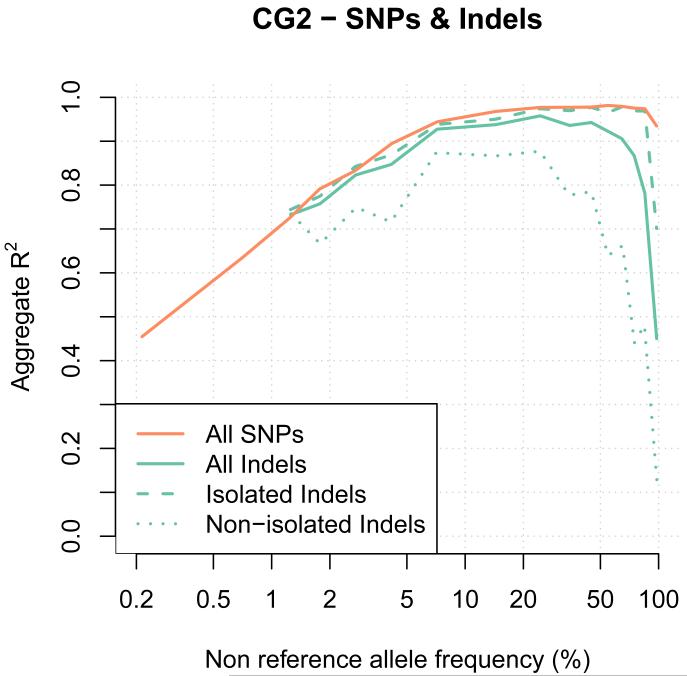
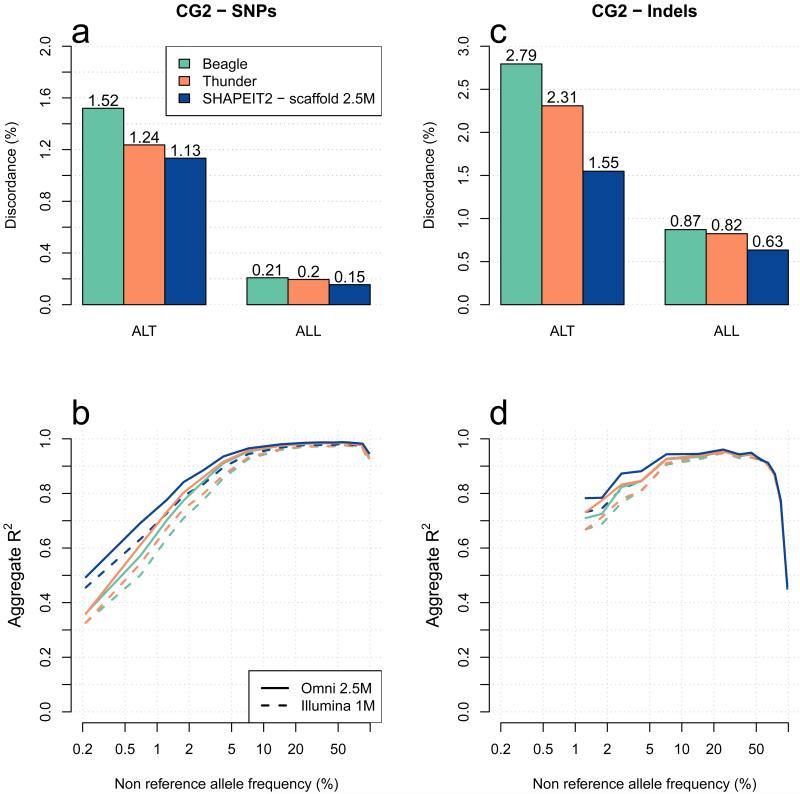
A major use of the 1000 Genomes Project (1000 GP) data is genotype imputation in genome-wide association studies (GWAS). Here we develop a method to estimate haplotypes from low-coverage sequencing data that can take advantage of single-nucleotide polymorphism (SNP) microarray genotypes on the same samples. First the SNP array data are phased to build a backbone (or 'scaffold') of haplotypes across each chromosome. We then phase the sequence data 'onto' this haplotype scaffold. This approach can take advantage of relatedness between sequenced and non-sequenced samples to improve accuracy. We use this method to create a new 1000 GP haplotype reference set for use by the human genetic community. Using a set of validation genotypes at SNP and bi-allelic indels we show that these haplotypes have lower genotype discordance and improved imputation performance into downstream GWAS samples, especially at low-frequency variants.
千人基因组计划(1000GP)数据的一个主要用途是在全基因组关联研究(GWAS)中进行基因型插补。在此,我们开发了一种从低覆盖度测序数据中估计单倍型的方法,该方法可以利用同一样本上的单核苷酸多态性(SNP)微阵列基因型。首先,对SNP阵列数据进行定相,以构建每条染色体上的单倍型主干(或“支架”)。然后,将序列数据“定相到”这个单倍型支架上。这种方法可以利用已测序样本和未测序样本之间的相关性来提高准确性。我们使用这种方法创建了一个新的1000GP单倍型参考集,供人类遗传学界使用。通过一组SNP和双等位基因插入缺失的验证基因型,我们表明这些单倍型具有较低的基因型不一致性,并提高了对下游GWAS样本的插补性能,尤其是在低频变异方面。