Department of Physiology, School of Medicine, Johns Hopkins University Baltimore, MD, USA.
Front Genet. 2015 Feb 3;6:3. doi: 10.3389/fgene.2015.00003. eCollection 2015.
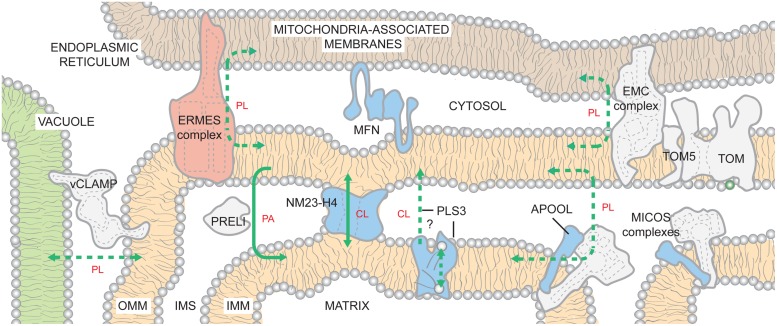
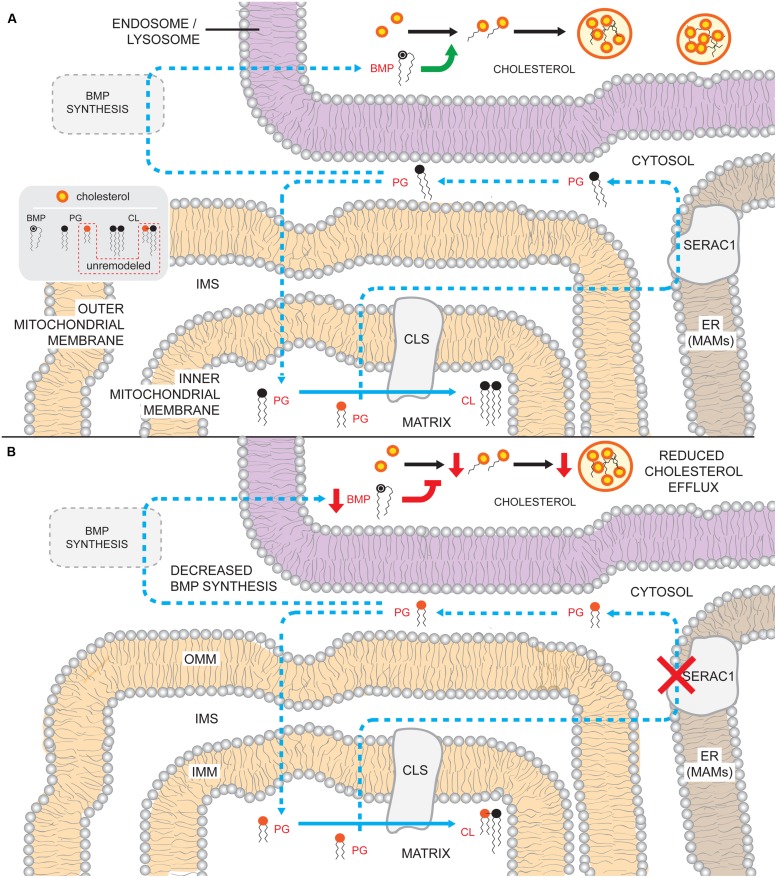
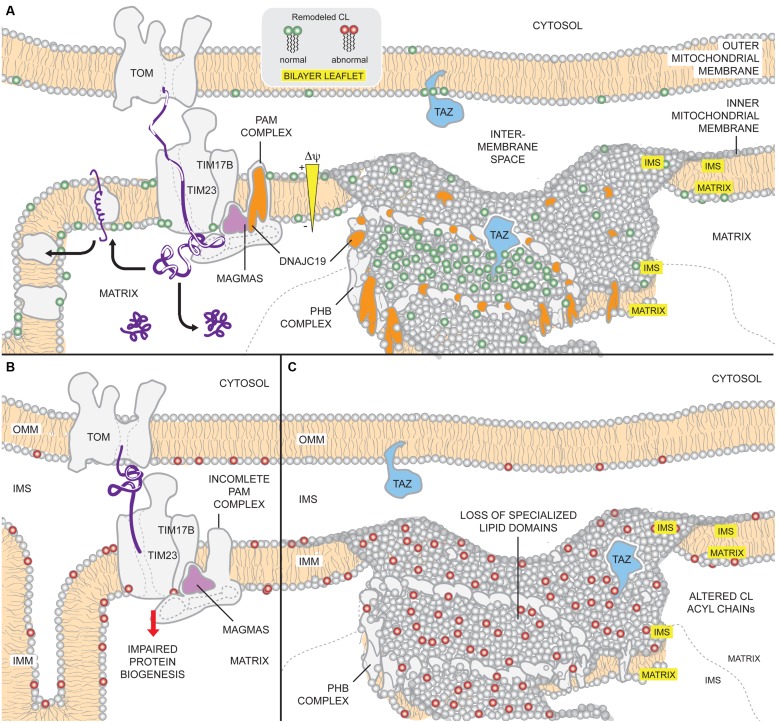
The human nuclear and mitochondrial genomes co-exist within each cell. While the mitochondrial genome encodes for a limited number of proteins, transfer RNAs, and ribosomal RNAs, the vast majority of mitochondrial proteins are encoded in the nuclear genome. Of the multitude of mitochondrial disorders known to date, only a fifth are maternally inherited. The recent characterization of the mitochondrial proteome therefore serves as an important step toward delineating the nosology of a large spectrum of phenotypically heterogeneous diseases. Following the identification of the first nuclear gene defect to underlie a mitochondrial disorder, a plenitude of genetic variants that provoke mitochondrial pathophysiology have been molecularly elucidated and classified into six categories that impact: (1) oxidative phosphorylation (subunits and assembly factors); (2) mitochondrial DNA maintenance and expression; (3) mitochondrial protein import and assembly; (4) mitochondrial quality control (chaperones and proteases); (5) iron-sulfur cluster homeostasis; and (6) mitochondrial dynamics (fission and fusion). Here, we propose that an additional class of genetic variant be included in the classification schema to acknowledge the role of genetic defects in phospholipid biosynthesis, remodeling, and metabolism in mitochondrial pathophysiology. This seventh class includes a small but notable group of nuclear-encoded proteins whose dysfunction impacts normal mitochondrial phospholipid metabolism. The resulting human disorders present with a diverse array of pathologic consequences that reflect the variety of functions that phospholipids have in mitochondria and highlight the important role of proper membrane homeostasis in mitochondrial biology.
人类核基因组和线粒体基因组共同存在于每个细胞中。虽然线粒体基因组编码了数量有限的蛋白质、转移 RNA 和核糖体 RNA,但绝大多数线粒体蛋白是由核基因组编码的。迄今为止已知的众多线粒体疾病中,只有五分之一是母系遗传的。因此,最近对线粒体蛋白质组的描述是朝着阐明大量表型异质性疾病的分类学迈出的重要一步。在确定第一个导致线粒体疾病的核基因突变后,大量引起线粒体病理生理学的遗传变异已被分子阐明,并分为六类,分别影响:(1)氧化磷酸化(亚基和组装因子);(2)线粒体 DNA 维持和表达;(3)线粒体蛋白的输入和组装;(4)线粒体质量控制(伴侣蛋白和蛋白酶);(5)铁硫簇稳态;(6)线粒体动力学(分裂和融合)。在这里,我们建议在分类方案中增加一类遗传变异,以承认遗传缺陷在磷脂生物合成、重塑和代谢中的作用,从而影响线粒体病理生理学。这第七类包括一小部分但很显著的一组核编码蛋白,其功能障碍会影响正常的线粒体磷脂代谢。由此产生的人类疾病表现出多种多样的病理后果,反映了磷脂在线粒体中的多种功能,并强调了适当的膜动态平衡在线粒体生物学中的重要作用。