Song Jimin, Chen Kevin C
Genome Biol. 2015 Feb 12;16(1):33. doi: 10.1186/s13059-015-0598-0.
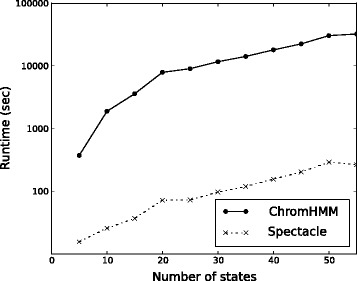
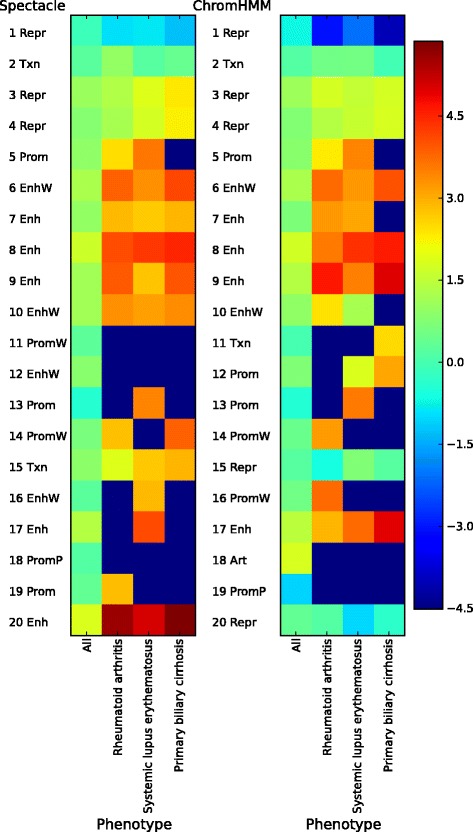
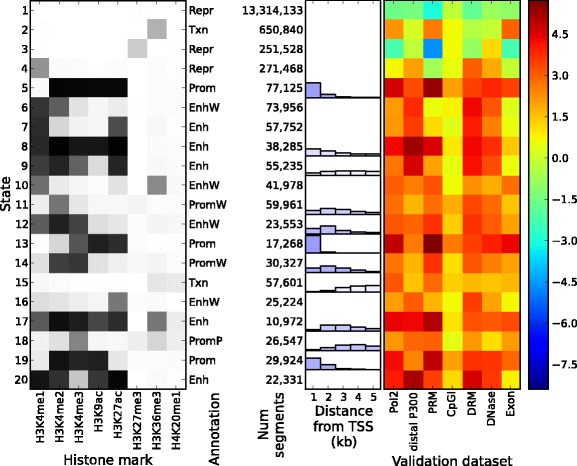
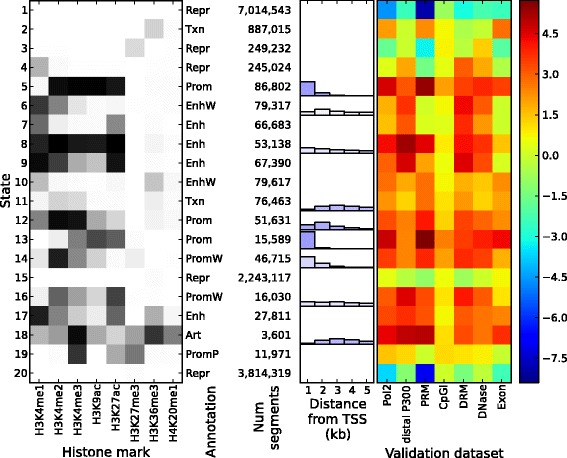
Epigenomic data from ENCODE can be used to associate specific combinations of chromatin marks with regulatory elements in the human genome. Hidden Markov models and the expectation-maximization (EM) algorithm are often used to analyze epigenomic data. However, the EM algorithm can have overfitting problems in data sets where the chromatin states show high class-imbalance and it is often slow to converge. Here we use spectral learning instead of EM and find that our software Spectacle overcame these problems. Furthermore, Spectacle is able to find enhancer subtypes not found by ChromHMM but strongly enriched in GWAS SNPs. Spectacle is available at https://github.com/jiminsong/Spectacle.
来自ENCODE的表观基因组数据可用于将特定的染色质标记组合与人类基因组中的调控元件相关联。隐马尔可夫模型和期望最大化(EM)算法常用于分析表观基因组数据。然而,在染色质状态显示出高度类别不平衡的数据集里,EM算法可能会出现过拟合问题,并且其收敛速度通常较慢。在这里,我们使用谱学习而非EM算法,发现我们的软件Spectacle克服了这些问题。此外,Spectacle能够找到ChromHMM未发现但在全基因组关联研究(GWAS)单核苷酸多态性(SNP)中高度富集的增强子亚型。可在https://github.com/jiminsong/Spectacle获取Spectacle。