Andres Marilou A, Cooke Ian M, Bellinger Frederick P, Berry Marla J, Zaporteza Maribel M, Rueli Rachel H, Barayuga Stephanie M, Chang Linda
Bekesy Laboratory of Neurobiology, Pacific Biosciences Research Center, University of Hawaii, Honolulu, Hawaii, USA.
Department of Biology, University of Hawaii, Honolulu, Hawaii, USA.
J Neurochem. 2015 Jul;134(1):56-65. doi: 10.1111/jnc.13104. Epub 2015 Apr 30.
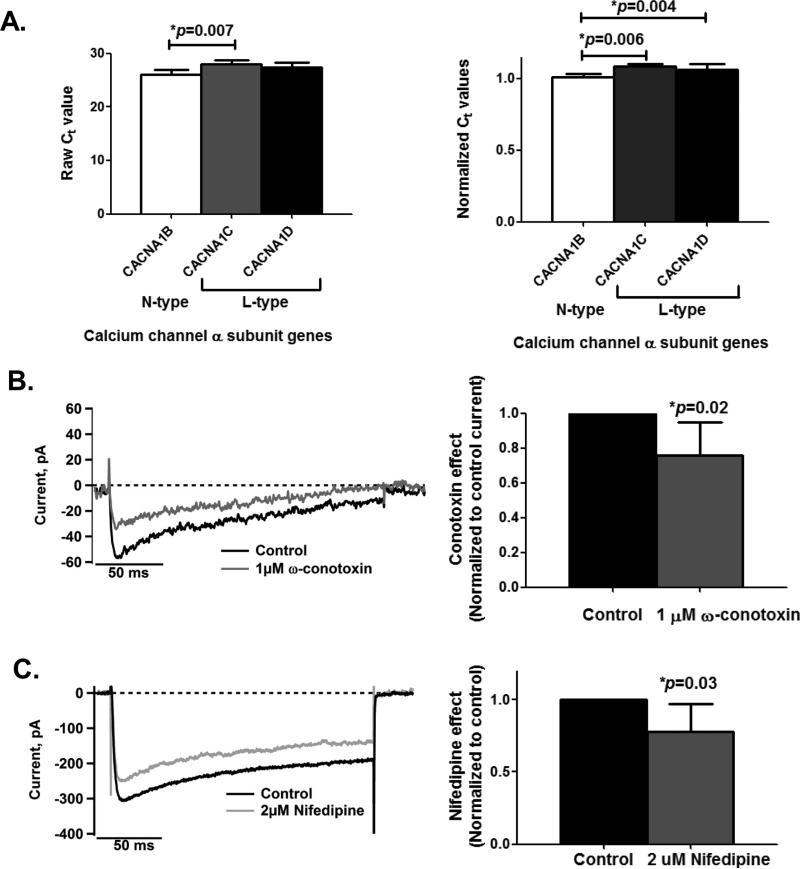
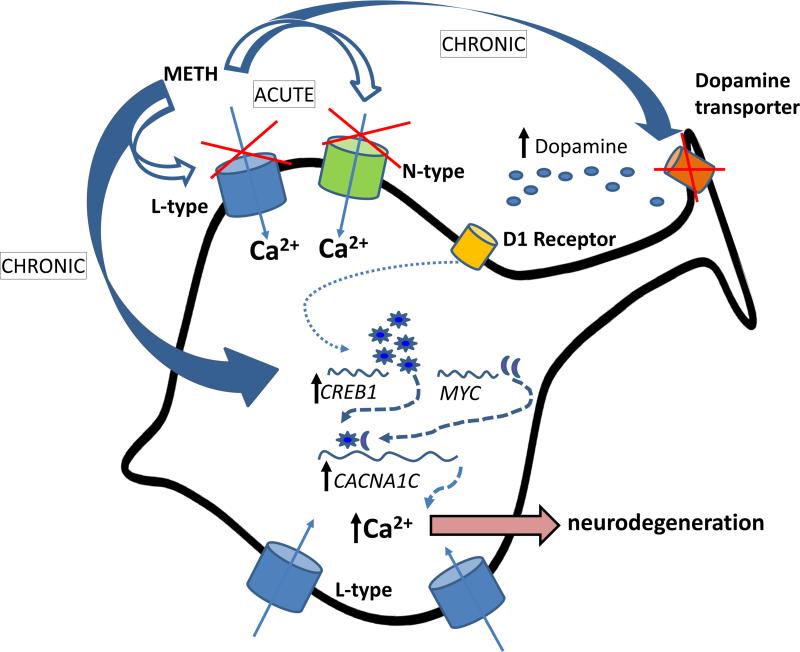
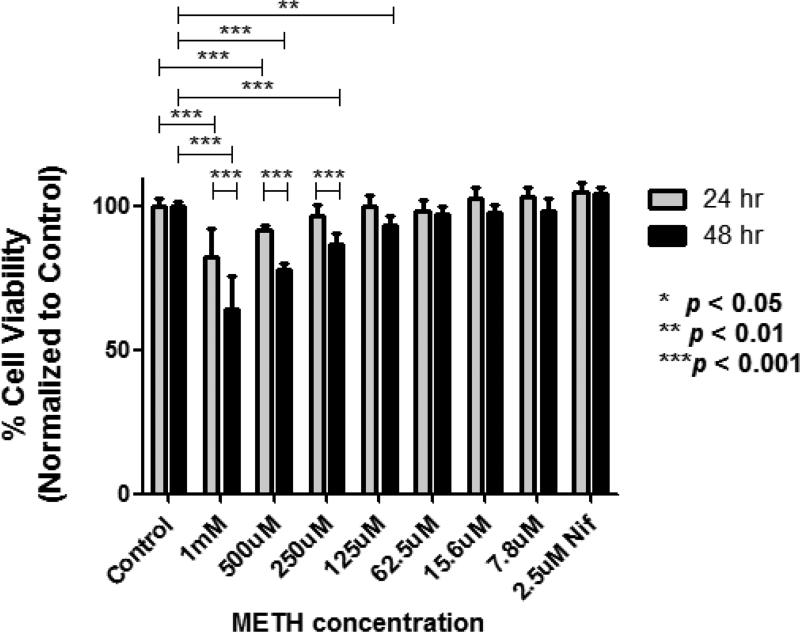
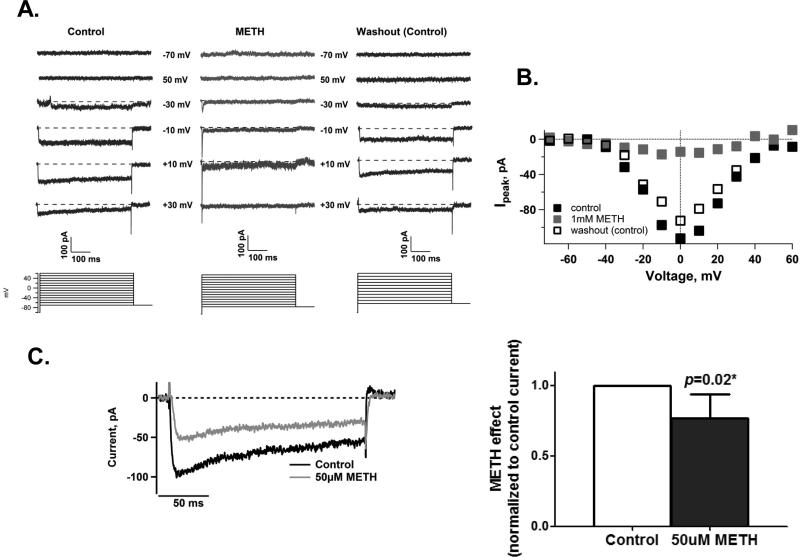
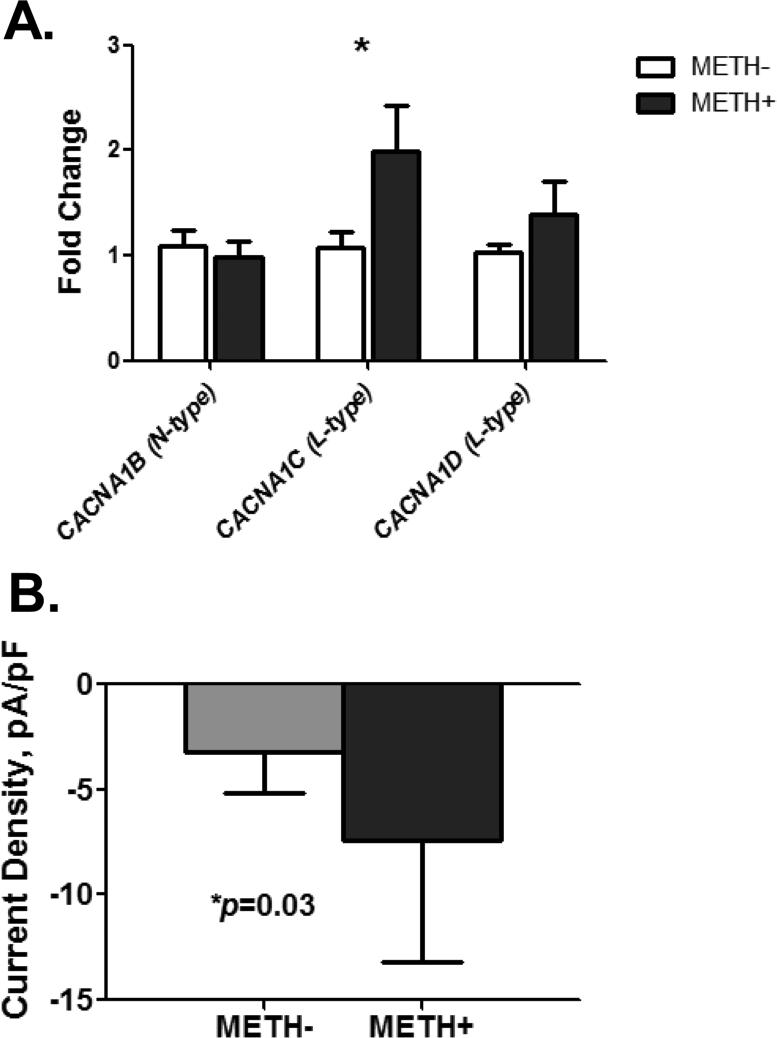
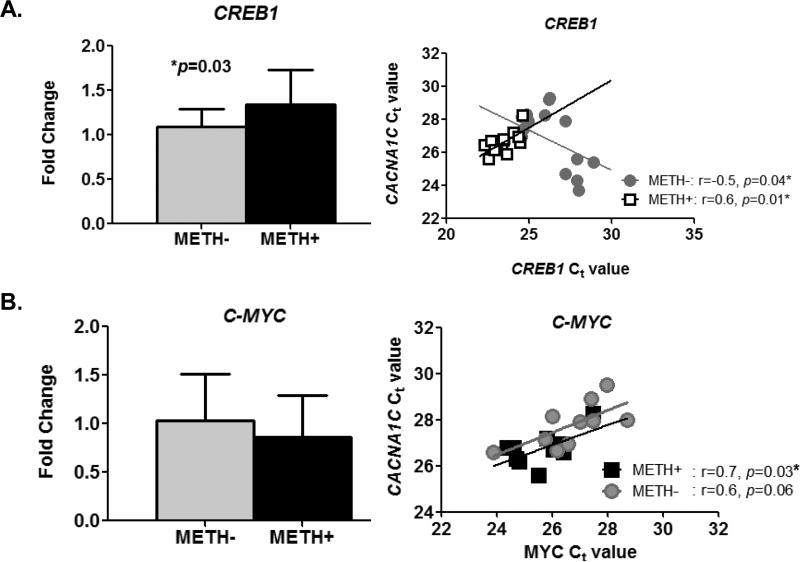
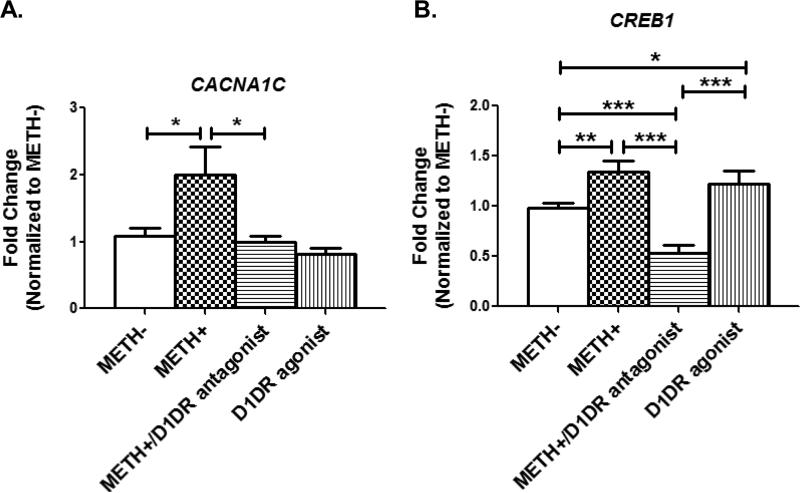
In neurons, calcium (Ca(2+) ) channels regulate a wide variety of functions ranging from synaptic transmission to gene expression. They also induce neuroplastic changes that alter gene expression following psychostimulant administration. Ca(2+) channel blockers have been considered as potential therapeutic agents for the treatment of methamphetamine (METH) dependence because of their ability to reduce drug craving among METH users. Here, we studied the effects of METH exposure on voltage-gated Ca(2+) channels using SH-SY5Y cells as a model of dopaminergic neurons. We found that METH has different short- and long-term effects. A short-term effect involves immediate (< 5 min) direct inhibition of Ca(2+) ion movements through Ca(2+) channels. Longer exposure to METH (20 min or 48 h) selectively up-regulates the expression of only the CACNA1C gene, thus increasing the number of L-type Ca(2+) channels. This up-regulation of CACNA1C is associated with the expression of the cAMP-responsive element-binding protein (CREB), a known regulator of CACNA1C gene expression, and the MYC gene, which encodes a transcription factor that putatively binds to a site proximal to the CACNA1C gene transcription initiation site. The short-term inhibition of Ca(2+) ion movement and later, the up-regulation of Ca(2+) channel gene expression together suggest the operation of cAMP-responsive element-binding protein- and C-MYC-mediated mechanisms to compensate for Ca(2+) channel inhibition by METH. Increased Ca(2+) current density and subsequent increased intracellular Ca(2+) may contribute to the neurodegeneration accompanying chronic METH abuse. Methamphetamine (METH) exposure has both short- and long-term effects. Acutely, methamphetamine directly inhibits voltage-gated calcium channels. Chronically, neurons compensate by up-regulating the L-type Ca(2+) channel gene, CACNA1C. This compensatory mechanism is mediated by transcription factors C-MYC and CREB, in which CREB is linked to the dopamine D1 receptor signaling pathway. These findings suggest Ca(2+) -mediated neurotoxicity owing to over-expression of calcium channels.
在神经元中,钙(Ca(2+))通道调节从突触传递到基因表达等多种功能。它们还会引发神经可塑性变化,这些变化会在给予精神兴奋剂后改变基因表达。由于钙通道阻滞剂能够减少甲基苯丙胺(METH)使用者对药物的渴望,因此被认为是治疗甲基苯丙胺成瘾的潜在治疗药物。在此,我们以SH-SY5Y细胞作为多巴胺能神经元模型,研究了METH暴露对电压门控钙通道的影响。我们发现METH具有不同的短期和长期效应。短期效应包括立即(<5分钟)直接抑制通过钙通道的钙离子移动。长时间暴露于METH(20分钟或48小时)会选择性地上调仅CACNA1C基因的表达,从而增加L型钙通道的数量。CACNA1C的这种上调与cAMP反应元件结合蛋白(CREB,一种已知的CACNA1C基因表达调节剂)以及MYC基因的表达相关,MYC基因编码一种转录因子,推测该转录因子与CACNA1C基因转录起始位点近端的一个位点结合。钙离子移动的短期抑制以及随后钙通道基因表达的上调共同表明存在由cAMP反应元件结合蛋白和C-MYC介导的机制,以补偿METH对钙通道的抑制。增加的钙电流密度以及随后细胞内钙的增加可能导致慢性METH滥用伴随的神经退行性变。甲基苯丙胺(METH)暴露具有短期和长期效应。急性情况下,甲基苯丙胺直接抑制电压门控钙通道。慢性情况下,神经元通过上调L型钙通道基因CACNA1C进行补偿。这种补偿机制由转录因子C-MYC和CREB介导,其中CREB与多巴胺D1受体信号通路相关。这些发现表明由于钙通道过度表达导致钙介导的神经毒性。