Lo Yu-Chen, Senese Silvia, Li Chien-Ming, Hu Qiyang, Huang Yong, Damoiseaux Robert, Torres Jorge Z
Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, California, United States of America; Program in Bioengineering, University of California, Los Angeles, Los Angeles, California, United States of America.
Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, California, United States of America.
PLoS Comput Biol. 2015 Mar 31;11(3):e1004153. doi: 10.1371/journal.pcbi.1004153. eCollection 2015 Mar.
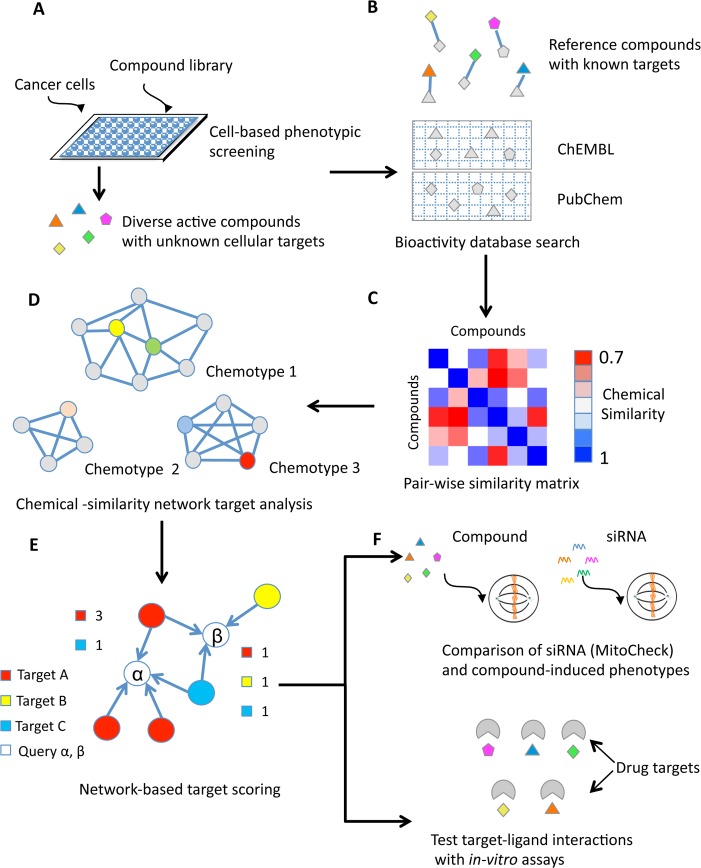
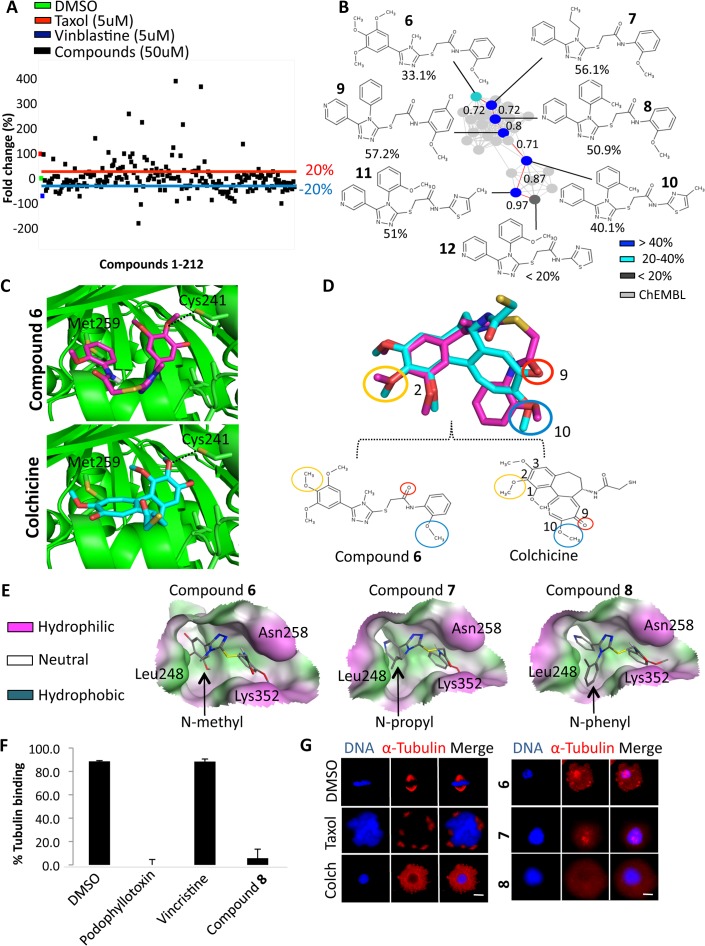
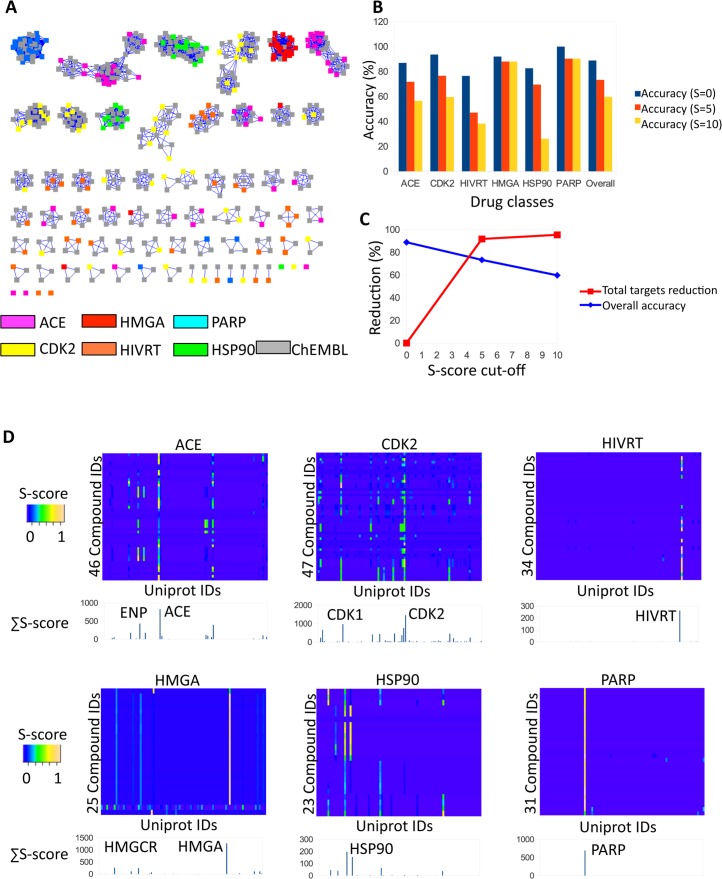
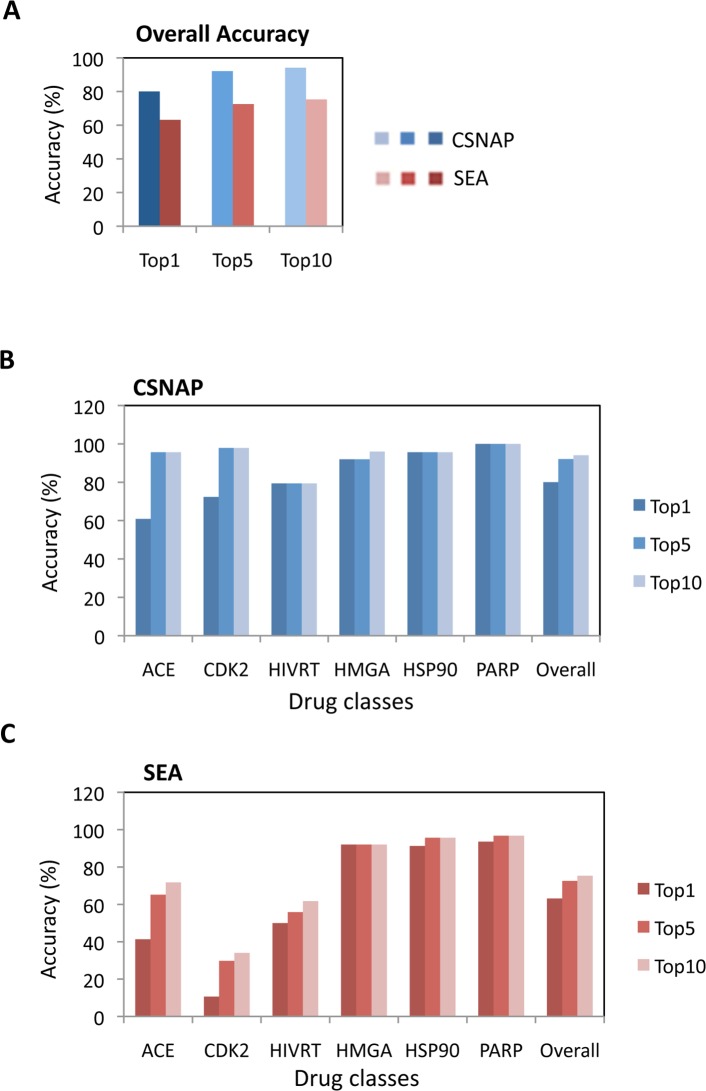
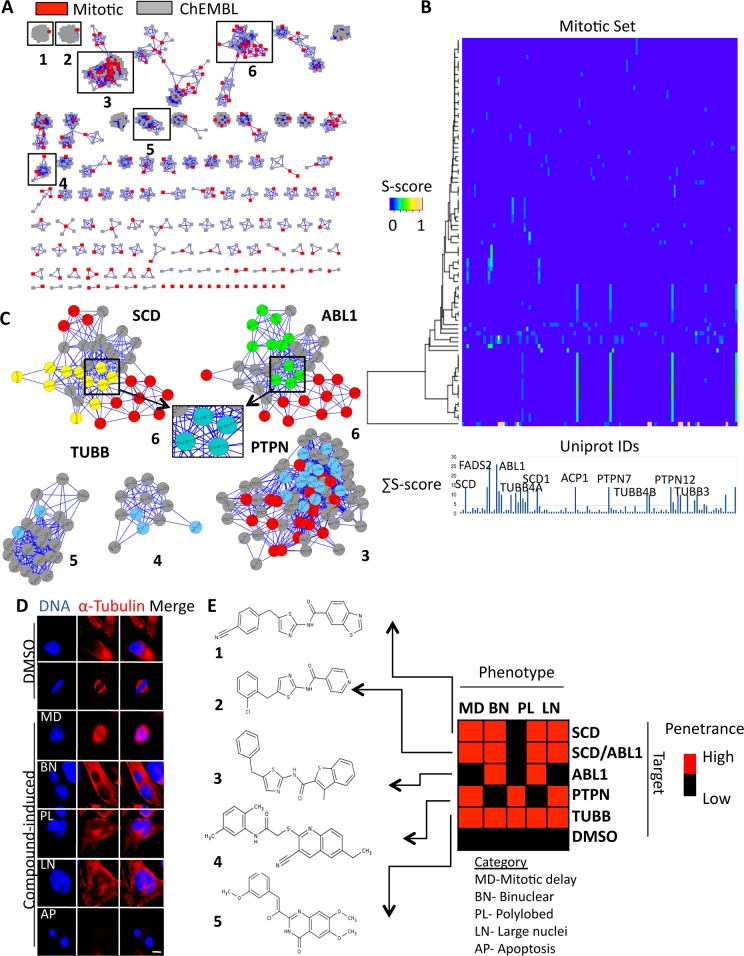
Target identification is one of the most critical steps following cell-based phenotypic chemical screens aimed at identifying compounds with potential uses in cell biology and for developing novel disease therapies. Current in silico target identification methods, including chemical similarity database searches, are limited to single or sequential ligand analysis that have limited capabilities for accurate deconvolution of a large number of compounds with diverse chemical structures. Here, we present CSNAP (Chemical Similarity Network Analysis Pulldown), a new computational target identification method that utilizes chemical similarity networks for large-scale chemotype (consensus chemical pattern) recognition and drug target profiling. Our benchmark study showed that CSNAP can achieve an overall higher accuracy (>80%) of target prediction with respect to representative chemotypes in large (>200) compound sets, in comparison to the SEA approach (60-70%). Additionally, CSNAP is capable of integrating with biological knowledge-based databases (Uniprot, GO) and high-throughput biology platforms (proteomic, genetic, etc) for system-wise drug target validation. To demonstrate the utility of the CSNAP approach, we combined CSNAP's target prediction with experimental ligand evaluation to identify the major mitotic targets of hit compounds from a cell-based chemical screen and we highlight novel compounds targeting microtubules, an important cancer therapeutic target. The CSNAP method is freely available and can be accessed from the CSNAP web server (http://services.mbi.ucla.edu/CSNAP/).
靶点识别是基于细胞的表型化学筛选之后最关键的步骤之一,此类筛选旨在识别在细胞生物学中有潜在用途的化合物以及开发新的疾病治疗方法。当前的计算机靶点识别方法,包括化学相似性数据库搜索,仅限于单一或顺序配体分析,对于准确解卷积大量具有不同化学结构的化合物的能力有限。在此,我们提出了CSNAP(化学相似性网络分析下拉法),这是一种新的计算靶点识别方法,它利用化学相似性网络进行大规模化学型(共识化学模式)识别和药物靶点分析。我们的基准研究表明,与SEA方法(60 - 70%)相比,对于大型(>200)化合物集中的代表性化学型,CSNAP在靶点预测方面能够实现总体更高的准确率(>80%)。此外,CSNAP能够与基于生物学知识的数据库(Uniprot、GO)和高通量生物学平台(蛋白质组学、遗传学等)整合,以进行系统层面的药物靶点验证。为了证明CSNAP方法的实用性,我们将CSNAP的靶点预测与实验性配体评估相结合,以识别基于细胞的化学筛选中命中化合物的主要有丝分裂靶点,并且我们重点介绍了靶向微管的新型化合物,微管是一个重要的癌症治疗靶点。CSNAP方法可免费获取,可从CSNAP网络服务器(http://services.mbi.ucla.edu/CSNAP/)访问。