Zhang Dong, Li Jingping, Compton Rosana O, Robertson Jon, Goff Valorie H, Epps Ethan, Kong Wenqian, Kim Changsoo, Paterson Andrew H
Plant Genome Mapping Laboratory, University of Georgia, Athens, Georgia 30602 Institute of Bioinformatics, University of Georgia, Athens, Georgia 30602.
Plant Genome Mapping Laboratory, University of Georgia, Athens, Georgia 30602.
G3 (Bethesda). 2015 Mar 31;5(6):1117-28. doi: 10.1534/g3.115.017590.
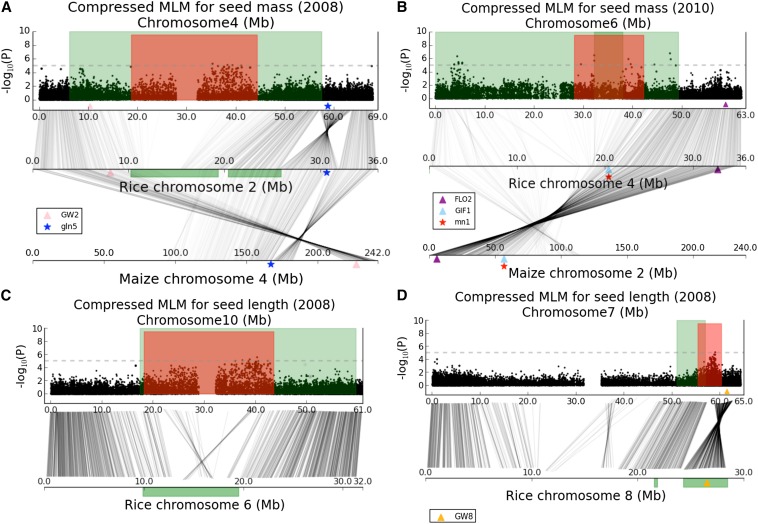
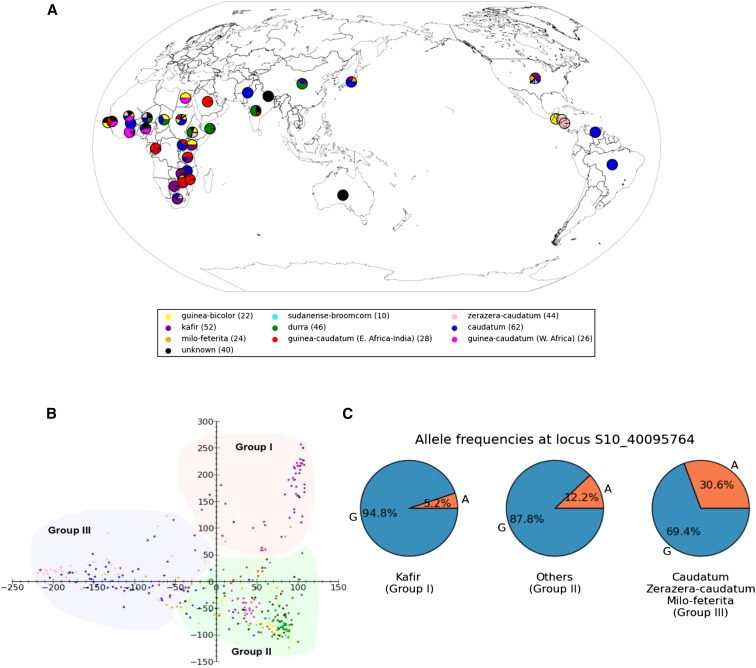
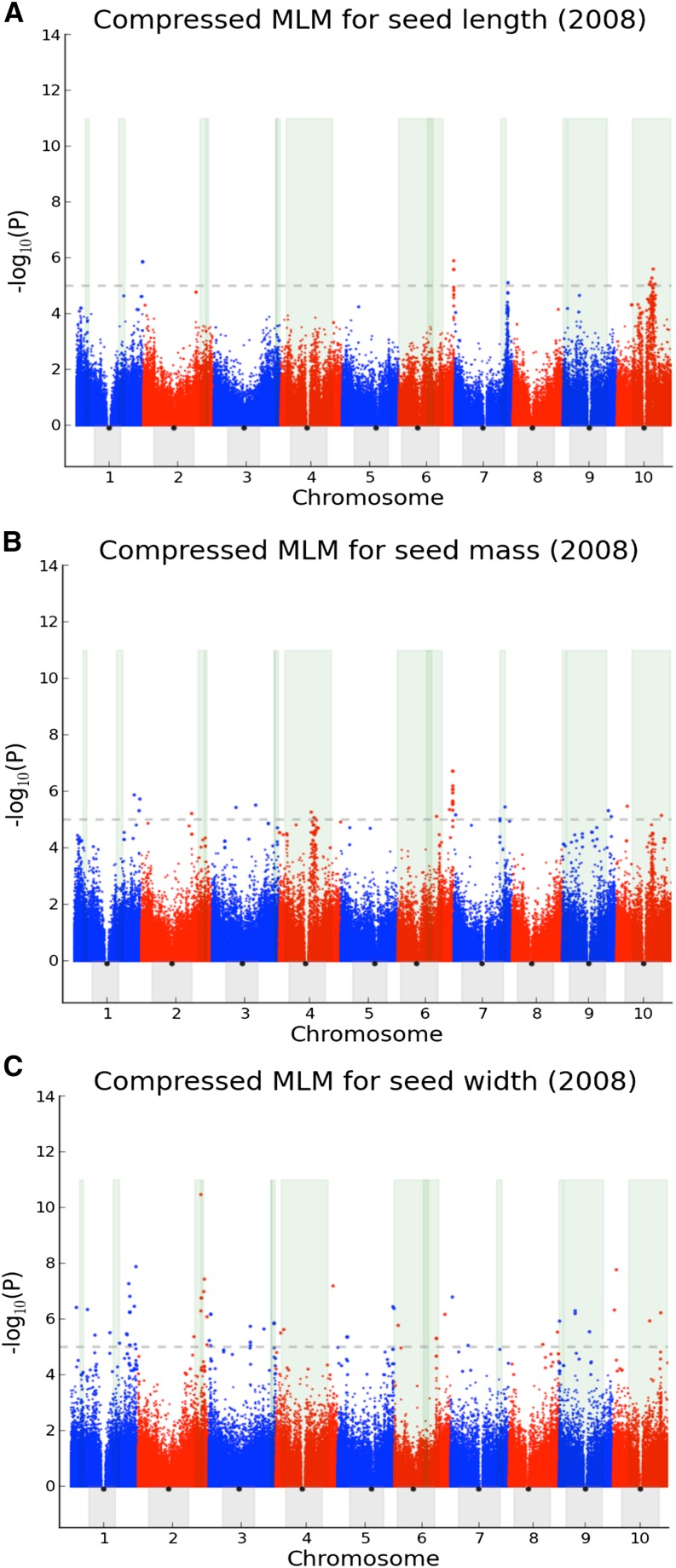
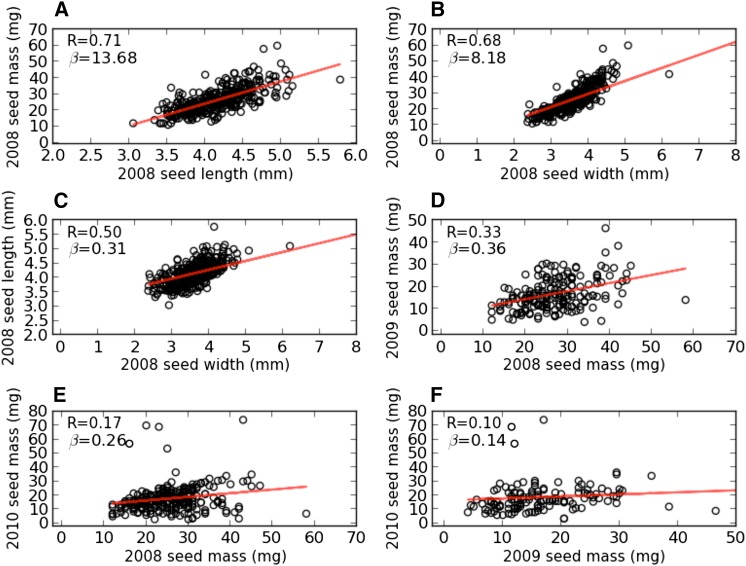
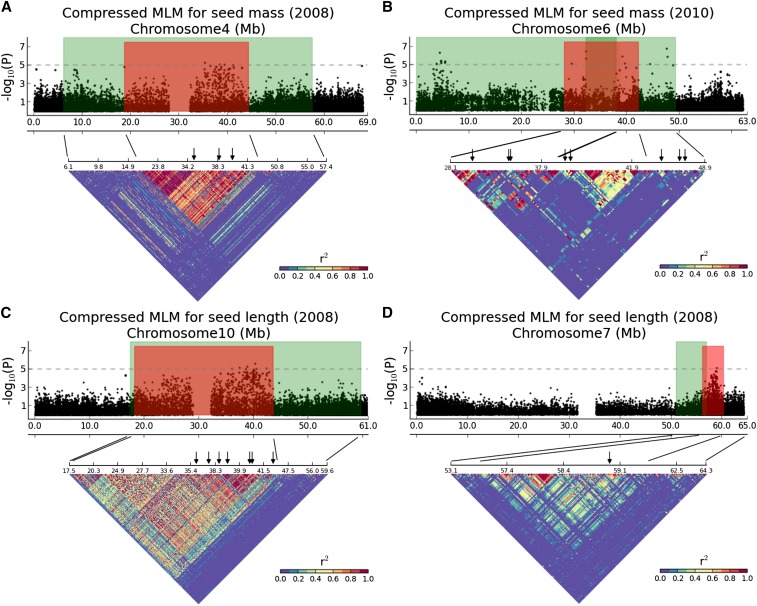
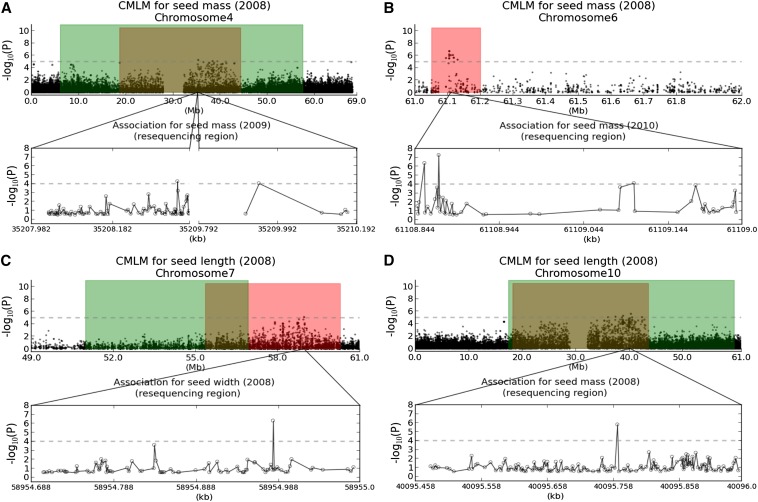
Seed size is closely related to fitness of wild plants, and its modification has been a key recurring element in domestication of seed/grain crops. In sorghum, a genomic and morphological model for panicoid cereals, a rich history of research into the genetics of seed size is reflected by a total of 13 likelihood intervals determined by conventional QTL (linkage) mapping in 11 nonoverlapping regions of the genome. To complement QTL data and investigate whether the discovery of seed size QTL is approaching "saturation," we compared QTL data to GWAS for seed mass, seed length, and seed width studied in 354 accessions from a sorghum association panel (SAP) that have been genotyped at 265,487 SNPs. We identified nine independent GWAS-based "hotspots" for seed size associations. Targeted resequencing near four association peaks with the most notable linkage disequilibrium provides further support of the role(s) of these regions in the genetic control of sorghum seed size and identifies two candidate causal variants with nonsynonymous mutations. Of nine GWAS hotspots in sorghum, seven have significant correspondence with rice QTL intervals and known genes for components of seed size on orthologous chromosomes. Identifying intersections between positional and association genetic data are a potentially powerful means to mitigate constraints associated with each approach, and nonrandom correspondence of sorghum (panicoid) GWAS signals to rice (oryzoid) QTL adds a new dimension to the ability to leverage genetic data about this important trait across divergent plants.
种子大小与野生植物的适应性密切相关,其改良一直是种子/谷物作物驯化过程中反复出现的关键因素。在高粱(一种黍族谷类作物的基因组和形态学模型)中,对种子大小遗传学的丰富研究历史体现在通过传统QTL(连锁)定位在基因组的11个非重叠区域确定的总共13个似然区间上。为了补充QTL数据并研究种子大小QTL的发现是否接近“饱和”,我们将QTL数据与在一个高粱关联群体(SAP)的354份材料中研究的种子重量、种子长度和种子宽度的全基因组关联研究(GWAS)数据进行了比较,这些材料在265,487个单核苷酸多态性(SNP)位点进行了基因分型。我们鉴定出了9个基于GWAS的种子大小关联“热点”。对四个具有最显著连锁不平衡的关联峰附近进行靶向重测序,进一步支持了这些区域在高粱种子大小遗传控制中的作用,并鉴定出两个具有非同义突变的候选因果变异。在高粱的9个GWAS热点中,有7个与水稻QTL区间以及直系同源染色体上种子大小组成部分的已知基因有显著对应关系。确定定位遗传数据和关联遗传数据之间的交集是减轻与每种方法相关的限制的一种潜在有力手段,并且高粱(黍族)GWAS信号与水稻(稻族)QTL的非随机对应为跨不同植物利用关于这一重要性状的遗传数据的能力增添了新的维度。