Rogers Matthew B, Song Timothy, Sebra Robert, Greenbaum Benjamin D, Hamelin Marie-Eve, Fitch Adam, Twaddle Alan, Cui Lijia, Holmes Edward C, Boivin Guy, Ghedin Elodie
Department of Computational and Systems Biology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
mBio. 2015 Apr 7;6(2):e02464-14. doi: 10.1128/mBio.02464-14.
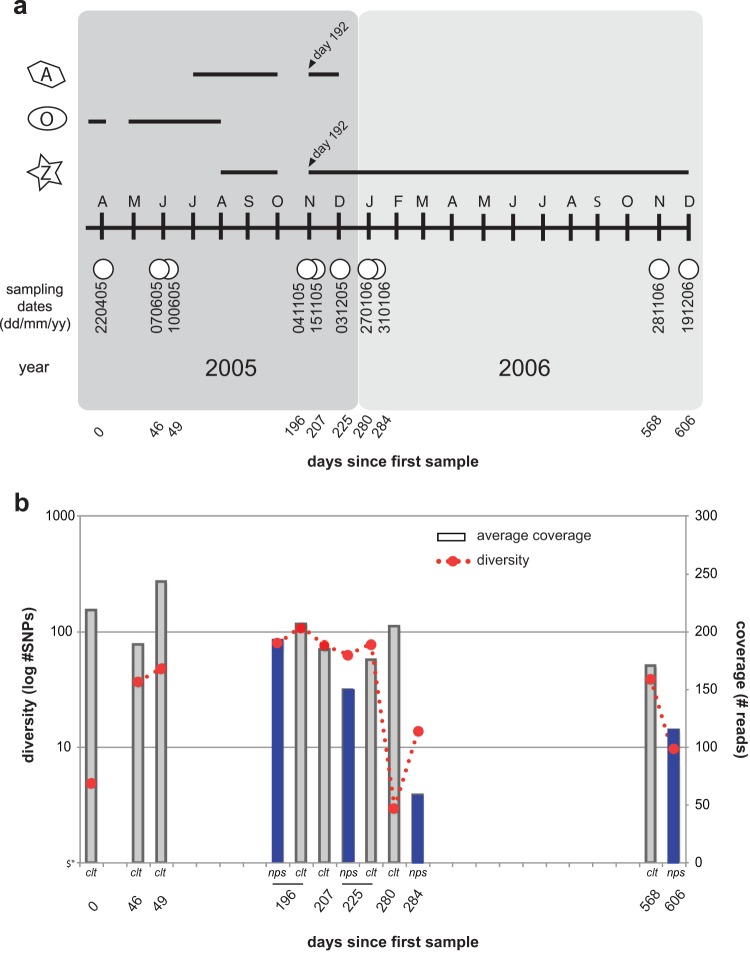
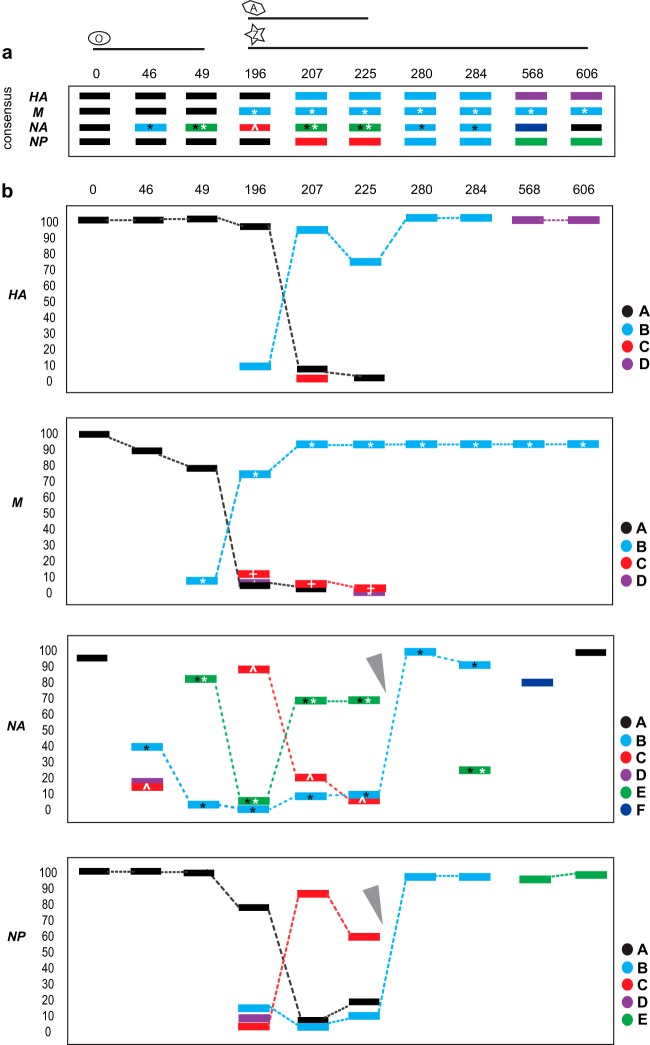
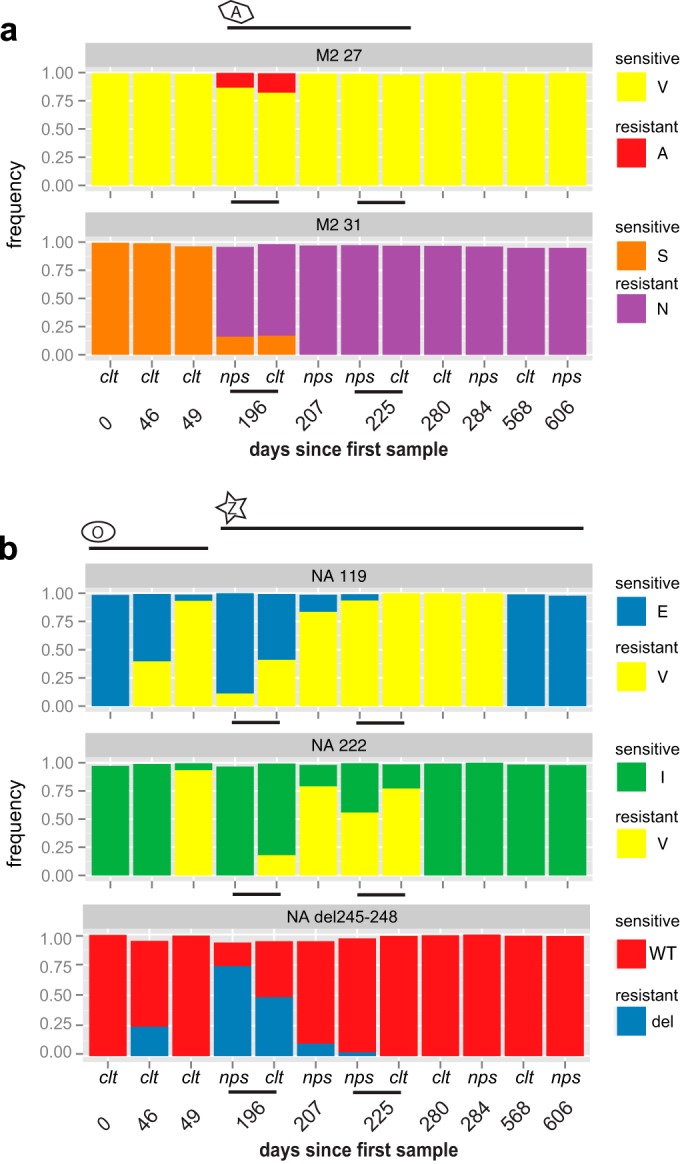
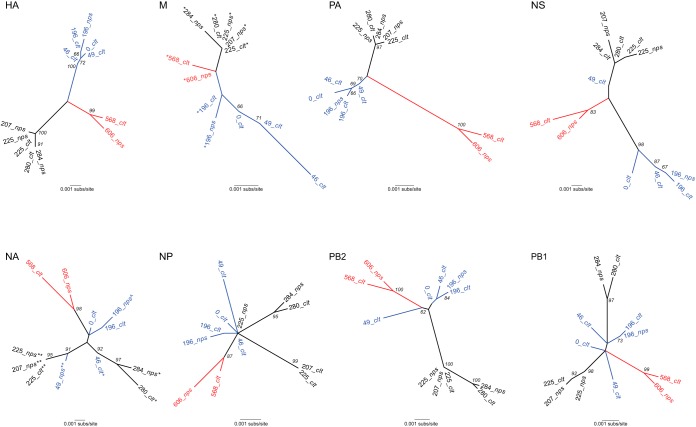
Resistance following antiviral therapy is commonly observed in human influenza viruses. Although this evolutionary process is initiated within individual hosts, little is known about the pattern, dynamics, and drivers of antiviral resistance at this scale, including the role played by reassortment. In addition, the short duration of human influenza virus infections limits the available time window in which to examine intrahost evolution. Using single-molecule sequencing, we mapped, in detail, the mutational spectrum of an H3N2 influenza A virus population sampled from an immunocompromised patient who shed virus over a 21-month period. In this unique natural experiment, we were able to document the complex dynamics underlying the evolution of antiviral resistance. Individual resistance mutations appeared weeks before they became dominant, evolved independently on cocirculating lineages, led to a genome-wide reduction in genetic diversity through a selective sweep, and were placed into new combinations by reassortment. Notably, despite frequent reassortment, phylogenetic analysis also provided evidence for specific patterns of segment linkage, with a strong association between the hemagglutinin (HA)- and matrix (M)-encoding segments that matches that previously observed at the epidemiological scale. In sum, we were able to reveal, for the first time, the complex interaction between multiple evolutionary processes as they occur within an individual host.
Understanding the evolutionary forces that shape the genetic diversity of influenza virus is crucial for predicting the emergence of drug-resistant strains but remains challenging because multiple processes occur concurrently. We characterized the evolution of antiviral resistance in a single persistent influenza virus infection, representing the first case in which reassortment and the complex patterns of drug resistance emergence and evolution have been determined within an individual host. Deep-sequence data from multiple time points revealed that the evolution of antiviral resistance reflects a combination of frequent mutation, natural selection, and a complex pattern of segment linkage and reassortment. In sum, these data show how immunocompromised hosts may help reveal the drivers of strain emergence.
抗病毒治疗后的耐药性在人类流感病毒中普遍存在。尽管这一进化过程在个体宿主内启动,但在这个尺度上,人们对抗病毒耐药性的模式、动态和驱动因素了解甚少,包括重配所起的作用。此外,人类流感病毒感染的持续时间较短,限制了研究宿主内进化的可用时间窗口。我们使用单分子测序技术,详细绘制了从一名免疫功能低下患者体内分离出的H3N2甲型流感病毒群体的突变谱,该患者在21个月的时间里持续排出病毒。在这个独特的自然实验中,我们能够记录抗病毒耐药性进化背后的复杂动态。个体耐药性突变在成为优势突变前数周就已出现,在同时传播的谱系中独立进化,通过选择性清除导致全基因组遗传多样性降低,并通过重配进入新的组合。值得注意的是,尽管频繁发生重配,但系统发育分析也为片段连锁的特定模式提供了证据,血凝素(HA)编码片段和基质(M)编码片段之间存在强关联,这与之前在流行病学尺度上观察到的情况相符。总之,我们首次揭示了多个进化过程在个体宿主内发生时的复杂相互作用。
了解塑造流感病毒遗传多样性的进化力量对于预测耐药菌株的出现至关重要,但由于多个过程同时发生,这仍然具有挑战性。我们对单一持续性流感病毒感染中的抗病毒耐药性进化进行了表征,这是首次在个体宿主内确定重配以及耐药性出现和进化的复杂模式。来自多个时间点的深度测序数据表明,抗病毒耐药性的进化反映了频繁突变、自然选择以及片段连锁和重配的复杂模式的组合。总之,这些数据表明免疫功能低下的宿主可能有助于揭示菌株出现的驱动因素。