Berenstein Ariel José, Piñero Janet, Furlong Laura Inés, Chernomoretz Ariel
Departamento de Física, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires and Instituto de Física de Buenos Aires, Consejo Nacional de Investigaciones Científicas y Técnicas, Pabellón 1, Ciudad Universitaria, Buenos Aires, Argentina.
Research Programme on Biomedical Informatics (GRIB), Hospital del Mar Medical Research Institute (IMIM), Universitat Pompeu Fabra (UPF), Carrer del Dr. Aiguader, 88, 08003-Barcelona, Spain.
PLoS One. 2015 Apr 9;10(4):e0122477. doi: 10.1371/journal.pone.0122477. eCollection 2015.
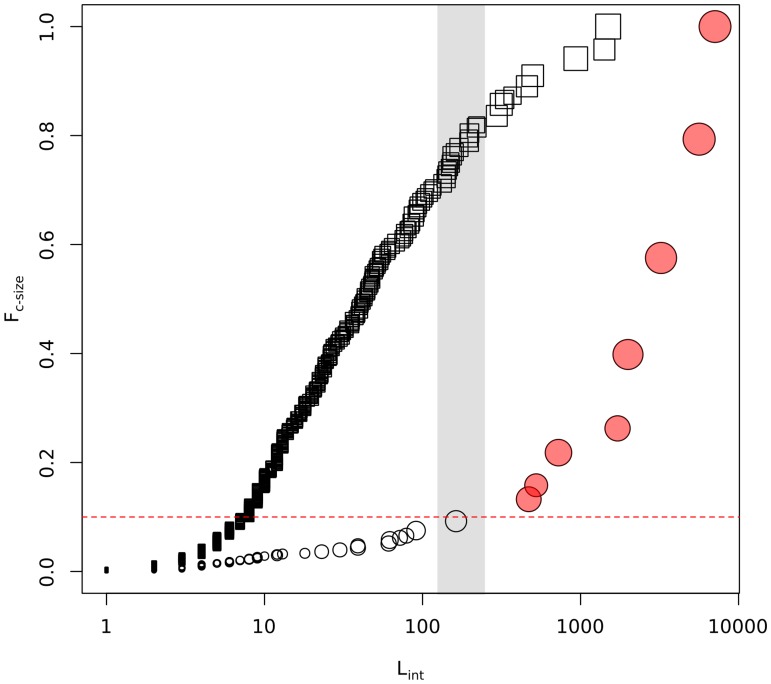
Cluster-based descriptions of biological networks have received much attention in recent years fostered by accumulated evidence of the existence of meaningful correlations between topological network clusters and biological functional modules. Several well-performing clustering algorithms exist to infer topological network partitions. However, due to respective technical idiosyncrasies they might produce dissimilar modular decompositions of a given network. In this contribution, we aimed to analyze how alternative modular descriptions could condition the outcome of follow-up network biology analysis.
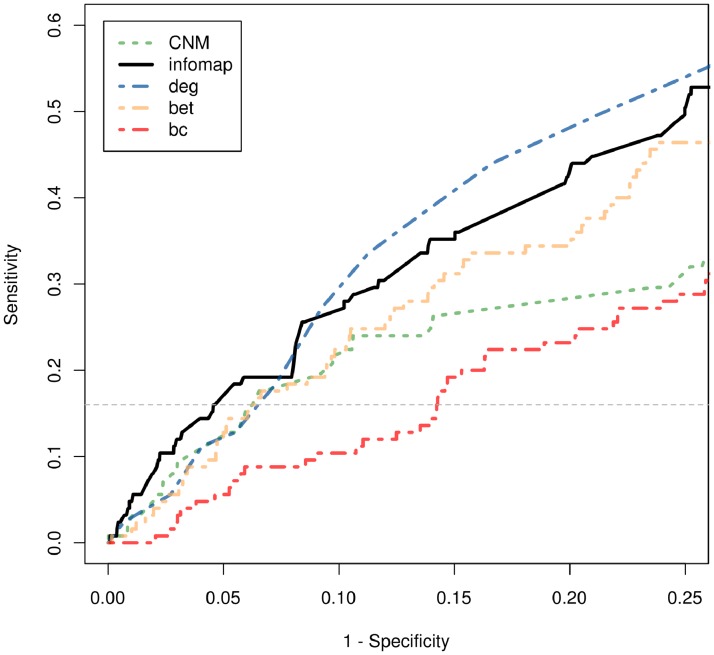
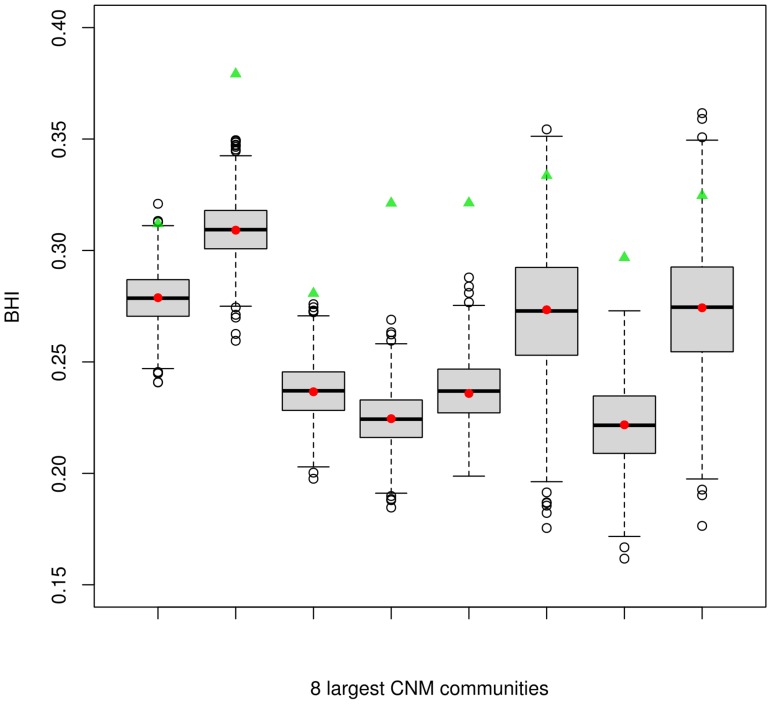
We considered a human protein interaction network and two paradigmatic cluster recognition algorithms, namely: the Clauset-Newman-Moore and the infomap procedures. We analyzed to what extent both methodologies yielded different results in terms of granularity and biological congruency. In addition, taking into account Guimera's cartographic role characterization of network nodes, we explored how the adoption of a given clustering methodology impinged on the ability to highlight relevant network meso-scale connectivity patterns.
As a case study we considered a set of aging related proteins and showed that only the high-resolution modular description provided by infomap, could unveil statistically significant associations between them and inter/intra modular cartographic features. Besides reporting novel biological insights that could be gained from the discovered associations, our contribution warns against possible technical concerns that might affect the tools used to mine for interaction patterns in network biology studies. In particular our results suggested that sub-optimal partitions from the strict point of view of their modularity levels might still be worth being analyzed when meso-scale features were to be explored in connection with external source of biological knowledge.
近年来,基于聚类的生物网络描述受到了广泛关注,这得益于拓扑网络聚类与生物功能模块之间存在有意义关联的累积证据。存在几种性能良好的聚类算法来推断拓扑网络分区。然而,由于各自的技术特性,它们可能会对给定网络产生不同的模块分解。在本研究中,我们旨在分析不同的模块描述如何影响后续网络生物学分析的结果。
我们考虑了一个人类蛋白质相互作用网络以及两种典型的聚类识别算法,即:克劳塞特 - 纽曼 - 摩尔算法和信息图算法。我们分析了这两种方法在粒度和生物学一致性方面产生不同结果的程度。此外,考虑到吉梅拉对网络节点的制图角色特征描述,我们探讨了采用给定的聚类方法如何影响突出相关网络中尺度连通性模式的能力。
作为一个案例研究,我们考虑了一组与衰老相关的蛋白质,并表明只有信息图提供的高分辨率模块描述能够揭示它们与模块间/模块内制图特征之间具有统计学意义的关联。除了报告从发现的关联中可以获得的新生物学见解外,我们的研究还警示了可能影响网络生物学研究中用于挖掘相互作用模式的工具的技术问题。特别是我们的结果表明,从模块性水平的严格角度来看次优的分区,在结合外部生物学知识源探索中尺度特征时,可能仍然值得分析。