Kukurba Kimberly R, Montgomery Stephen B
Department of Pathology, Stanford University School of Medicine, Stanford, California 94305; Department of Genetics, Stanford University School of Medicine, Stanford, California 94305;
Department of Pathology, Stanford University School of Medicine, Stanford, California 94305; Department of Genetics, Stanford University School of Medicine, Stanford, California 94305; Department of Computer Science, Stanford University School of Medicine, Stanford, California 94305.
Cold Spring Harb Protoc. 2015 Apr 13;2015(11):951-69. doi: 10.1101/pdb.top084970.
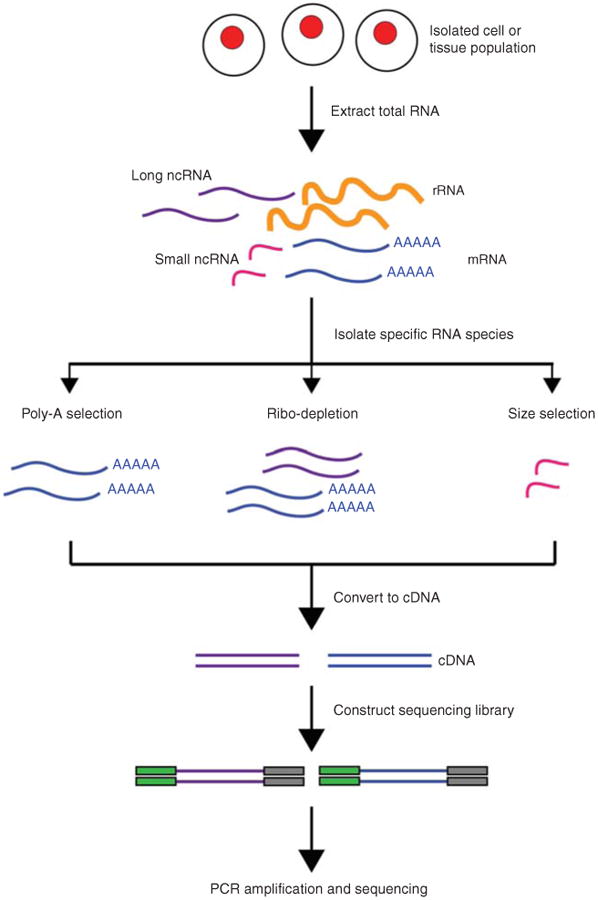
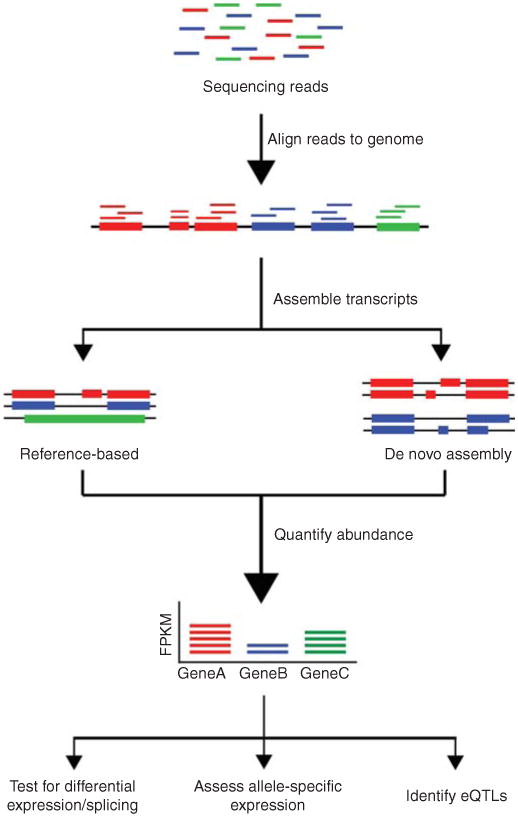
RNA sequencing (RNA-Seq) uses the capabilities of high-throughput sequencing methods to provide insight into the transcriptome of a cell. Compared to previous Sanger sequencing- and microarray-based methods, RNA-Seq provides far higher coverage and greater resolution of the dynamic nature of the transcriptome. Beyond quantifying gene expression, the data generated by RNA-Seq facilitate the discovery of novel transcripts, identification of alternatively spliced genes, and detection of allele-specific expression. Recent advances in the RNA-Seq workflow, from sample preparation to library construction to data analysis, have enabled researchers to further elucidate the functional complexity of the transcription. In addition to polyadenylated messenger RNA (mRNA) transcripts, RNA-Seq can be applied to investigate different populations of RNA, including total RNA, pre-mRNA, and noncoding RNA, such as microRNA and long ncRNA. This article provides an introduction to RNA-Seq methods, including applications, experimental design, and technical challenges.
RNA测序(RNA-Seq)利用高通量测序方法的能力来深入了解细胞的转录组。与以前基于桑格测序和微阵列的方法相比,RNA-Seq提供了更高的覆盖范围以及对转录组动态特性的更高分辨率。除了量化基因表达外,RNA-Seq产生的数据还有助于发现新的转录本、识别可变剪接基因以及检测等位基因特异性表达。从样品制备到文库构建再到数据分析,RNA-Seq工作流程的最新进展使研究人员能够进一步阐明转录的功能复杂性。除了聚腺苷酸化信使RNA(mRNA)转录本外,RNA-Seq还可用于研究不同的RNA群体,包括总RNA、前体mRNA和非编码RNA,如微小RNA和长链非编码RNA。本文介绍了RNA-Seq方法,包括应用、实验设计和技术挑战。