Nazem Amir, Sankowski Roman, Bacher Michael, Al-Abed Yousef
Elmezzi Graduate School of Molecular Medicine, The Feinstein Institute for Medical Research, 350 Community drive, Manhasset, NY, 11030, USA.
Institute of Immunology, Philipps University Marburg, Hans-Meerwein-Str., 35043, Marburg, Germany.
J Neuroinflammation. 2015 Apr 17;12:74. doi: 10.1186/s12974-015-0291-y.
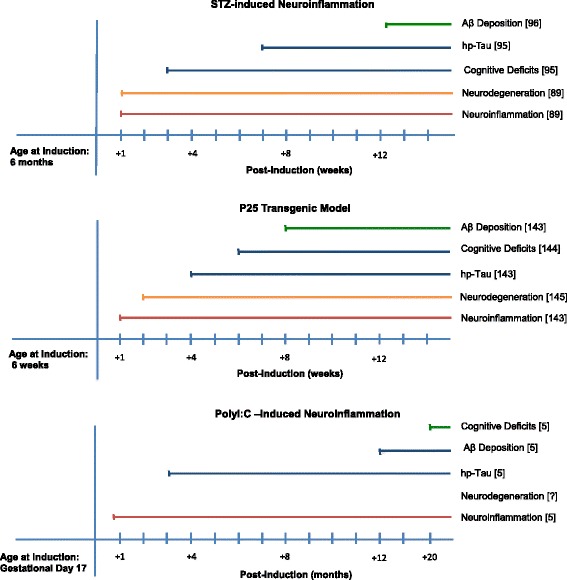
Alzheimer's disease remains incurable, and the failures of current disease-modifying strategies for Alzheimer's disease could be attributed to a lack of in vivo models that recapitulate the underlying etiology of late-onset Alzheimer's disease. The etiology of late-onset Alzheimer's disease is not based on mutations related to amyloid-β (Aβ) or tau production which are currently the basis of in vivo models of Alzheimer's disease. It has recently been suggested that mechanisms like chronic neuroinflammation may occur prior to amyloid-β and tau pathologies in late-onset Alzheimer's disease. The aim of this study is to analyze the characteristics of rodent models of neuroinflammation in late-onset Alzheimer's disease. Our search criteria were based on characteristics of an idealistic disease model that should recapitulate causes, symptoms, and lesions in a chronological order similar to the actual disease. Therefore, a model based on the inflammation hypothesis of late-onset Alzheimer's disease should include the following features: (i) primary chronic neuroinflammation, (ii) manifestations of memory and cognitive impairment, and (iii) late development of tau and Aβ pathologies. The following models fit the pre-defined criteria: lipopolysaccharide- and PolyI:C-induced models of immune challenge; streptozotocin-, okadaic acid-, and colchicine neurotoxin-induced neuroinflammation models, as well as interleukin-1β, anti-nerve growth factor and p25 transgenic models. Among these models, streptozotocin, PolyI:C-induced, and p25 neuroinflammation models are compatible with the inflammation hypothesis of Alzheimer's disease.
阿尔茨海默病仍然无法治愈,目前阿尔茨海默病疾病修饰策略的失败可能归因于缺乏能够重现晚发性阿尔茨海默病潜在病因的体内模型。晚发性阿尔茨海默病的病因并非基于与淀粉样蛋白-β(Aβ)或tau蛋白产生相关的突变,而目前这些突变是阿尔茨海默病体内模型的基础。最近有研究表明,在晚发性阿尔茨海默病中,慢性神经炎症等机制可能先于淀粉样蛋白-β和tau蛋白病变出现。本研究的目的是分析晚发性阿尔茨海默病神经炎症啮齿动物模型的特征。我们的搜索标准基于理想疾病模型的特征,该模型应按与实际疾病相似的时间顺序重现病因、症状和病变。因此,基于晚发性阿尔茨海默病炎症假说的模型应具备以下特征:(i)原发性慢性神经炎症;(ii)记忆和认知障碍表现;(iii)tau蛋白和Aβ蛋白病变的晚期发展。以下模型符合预先定义的标准:脂多糖和聚肌苷酸:聚胞苷酸诱导的免疫挑战模型;链脲佐菌素、冈田酸和秋水仙碱神经毒素诱导的神经炎症模型,以及白细胞介素-1β、抗神经生长因子和p25转基因模型。在这些模型中,链脲佐菌素、聚肌苷酸:聚胞苷酸诱导的和p25神经炎症模型与阿尔茨海默病的炎症假说相符。