Blacher Eran, Dadali Tulin, Bespalko Alina, Haupenthal Viola J, Grimm Marcus O W, Hartmann Tobias, Lund Frances E, Stein Reuven, Levy Ayelet
Department of Neurobiology, George S. Wise Faculty of Life Sciences, Tel Aviv University, Tel Aviv, Israel.
Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL.
Ann Neurol. 2015 Jul;78(1):88-103. doi: 10.1002/ana.24425. Epub 2015 May 25.
Alzheimer's disease (AD)-associated dementia is due to tissue damage caused by amyloid β (Aβ) deposition within the brain and by accompanying neuroinflammation. The nicotinamide adenine dinucleotide (NAD) glycohydrolase CD38, which is expressed by neurons, astrocytes, and microglial cells, regulates inflammatory and repair processes in the brain and other tissues by degrading NAD and repressing the activity of other NAD-consuming enzymes and by producing NAD-derived metabolites that regulate calcium signaling and migration of inflammatory cells. Given the role of CD38 in neuroinflammation and repair, we examined the effect of CD38 deletion on AD pathology.
We crossed APPswePS1ΔE9 (APP.PS) mice with Cd38(-) (/) (-) mice to generate AD-prone CD38-deficient animals (APP.PS.Cd38(-) (/) (-) ) and examined AD-related phenotypes in both groups.
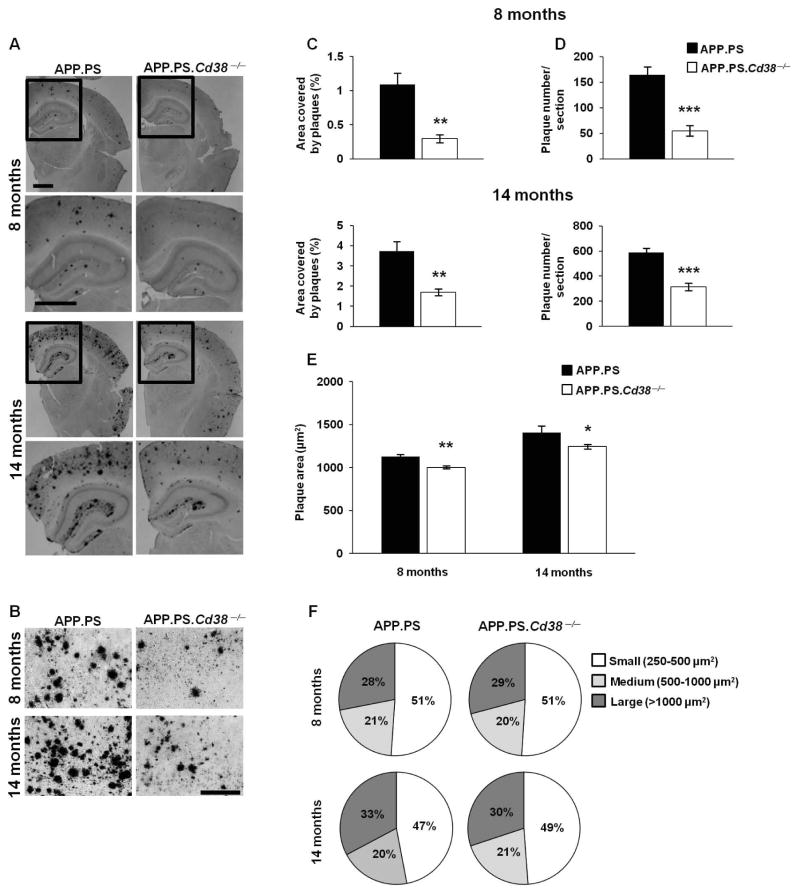
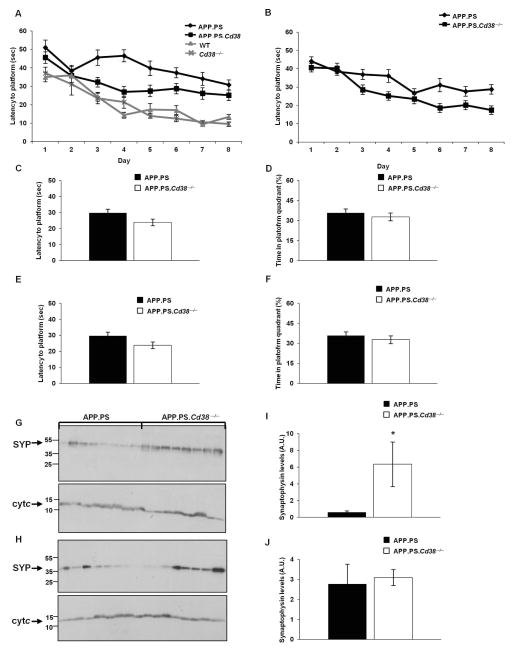
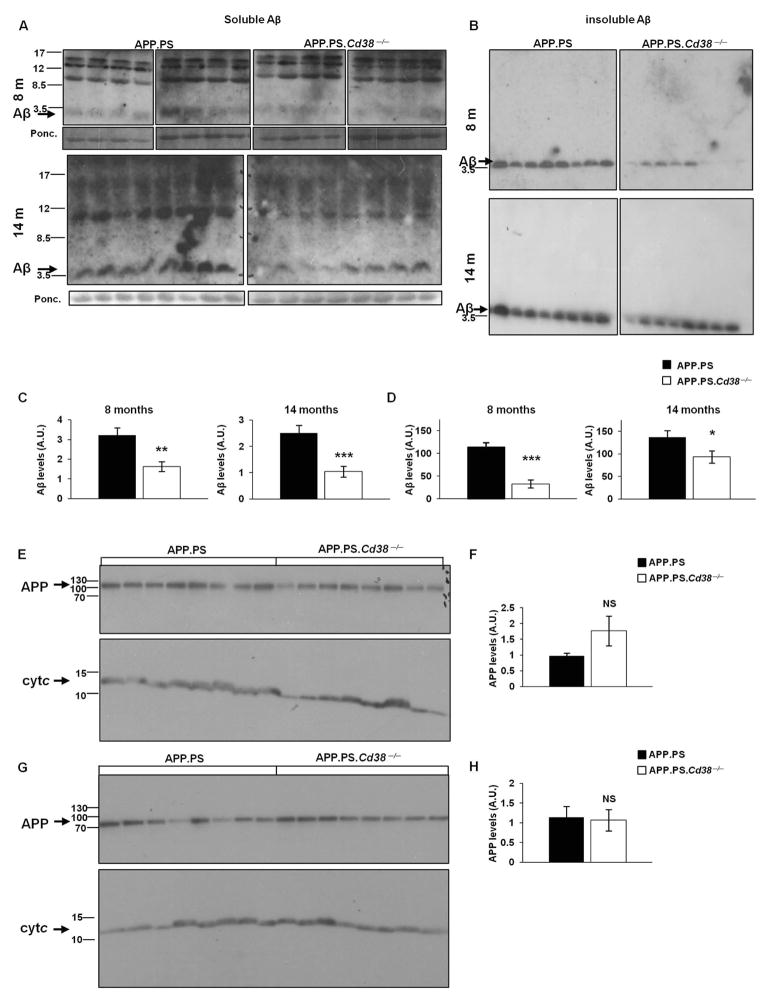
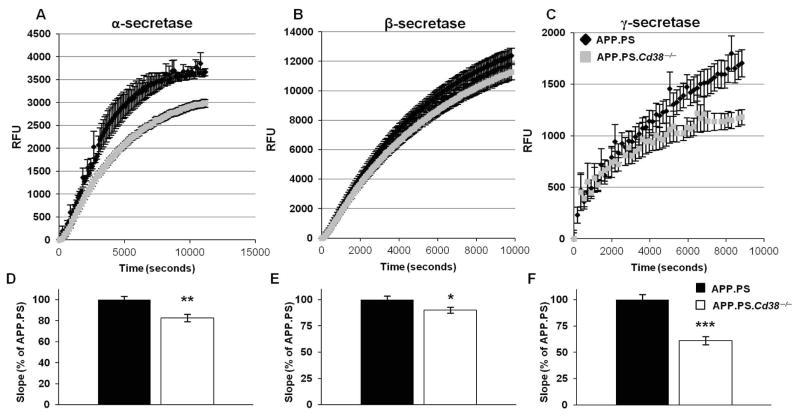
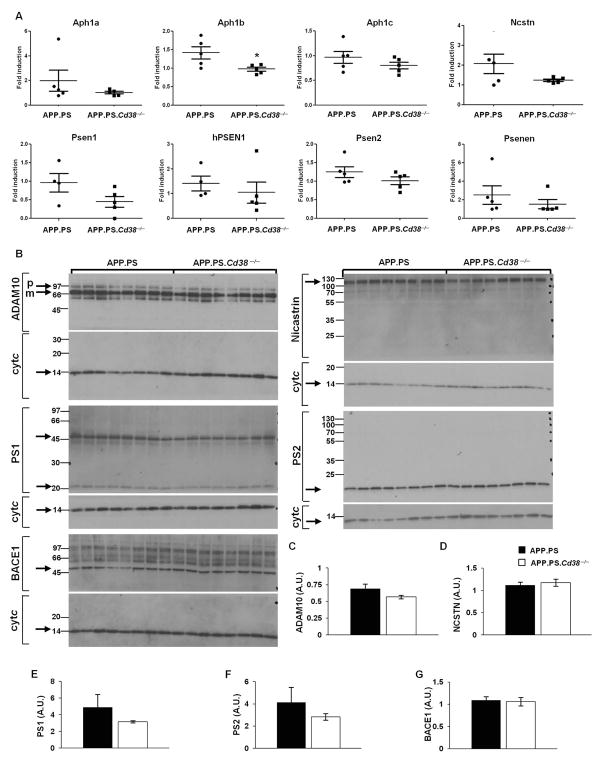
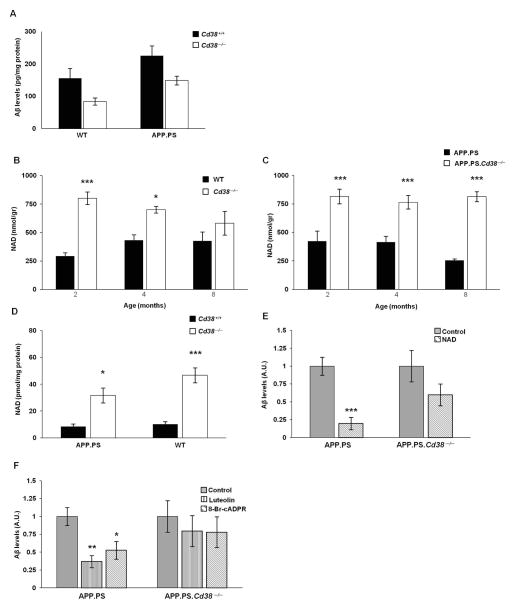
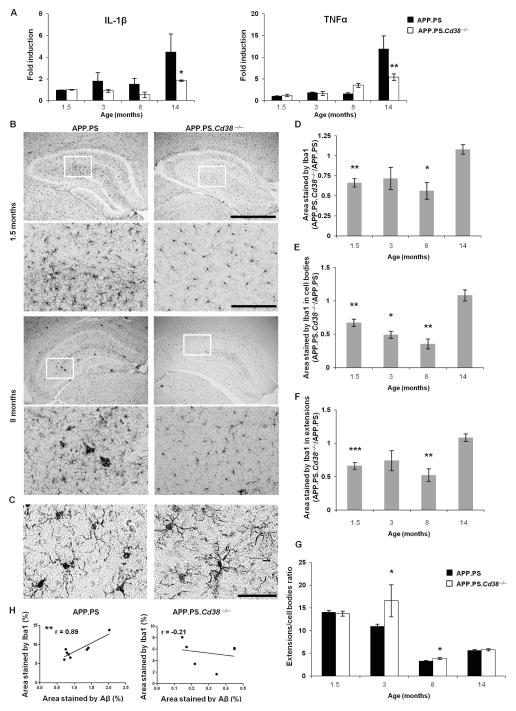
APP.PS.Cd38(-) (/) (-) mice exhibited significant reductions in Aβ plaque load and soluble Aβ levels compared to APP.PS mice, and this correlated with improved spatial learning. Although CD38 deficiency resulted in decreased microglia/macrophage (MM) accumulation, the transcription profile of the Cd38(-) (/) (-) and Cd38(+/) (+) MM was similar, suggesting that the decreased Aβ burden in APP.PS.Cd38(-) (/) (-) mice was not due to alterations in MM activation/function. Instead, APP.PS.Cd38(-) (/) (-) neuronal cultures secreted less Aβ and this reduction was mimicked when APP.PS neuronal cultures were treated with inhibitors that blocked CD38 enzyme activity or the signaling pathways controlled by CD38-derived metabolites. Furthermore, β- and γ-secretase activity was decreased in APP.PS.Cd38(-) (/) (-) mice, which correlated with decreased Aβ production.
CD38 regulates AD pathology in the APP.PS model of AD, suggesting that CD38 may be a novel target for AD treatment.
阿尔茨海默病(AD)相关痴呆是由淀粉样β蛋白(Aβ)在脑内沉积以及伴随的神经炎症引起的组织损伤所致。烟酰胺腺嘌呤二核苷酸(NAD)糖水解酶CD38由神经元、星形胶质细胞和小胶质细胞表达,通过降解NAD和抑制其他消耗NAD的酶的活性,以及产生调节钙信号和炎症细胞迁移的NAD衍生代谢物,来调节脑和其他组织中的炎症和修复过程。鉴于CD38在神经炎症和修复中的作用,我们研究了CD38基因缺失对AD病理的影响。
我们将APPswePS1ΔE9(APP.PS)小鼠与Cd38(- / -)小鼠杂交,以生成易患AD的CD38缺陷动物(APP.PS.Cd38(- / -)),并在两组中检查与AD相关的表型。
与APP.PS小鼠相比,APP.PS.Cd38(- / -)小鼠的Aβ斑块负荷和可溶性Aβ水平显著降低,这与空间学习能力的改善相关。虽然CD38缺陷导致小胶质细胞/巨噬细胞(MM)积累减少,但Cd38(- / -)和Cd38(+ / +) MM的转录谱相似,这表明APP.PS.Cd38(- / -)小鼠中Aβ负担的降低并非由于MM激活/功能的改变。相反,APP.PS.Cd38(- / -)神经元培养物分泌的Aβ较少,当用阻断CD38酶活性或由CD38衍生代谢物控制的信号通路的抑制剂处理APP.PS神经元培养物时,也会出现这种减少。此外,APP.PS.Cd38(- / -)小鼠中的β-和γ-分泌酶活性降低,这与Aβ产生减少相关。
CD38在AD的APP.PS模型中调节AD病理,提示CD38可能是AD治疗的新靶点。