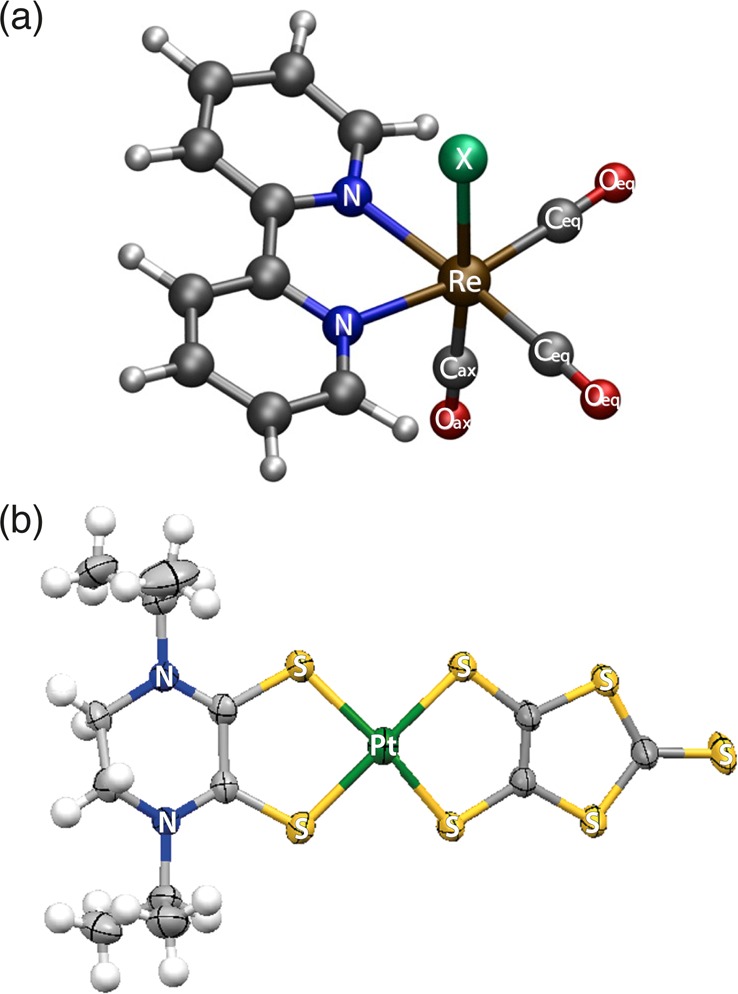
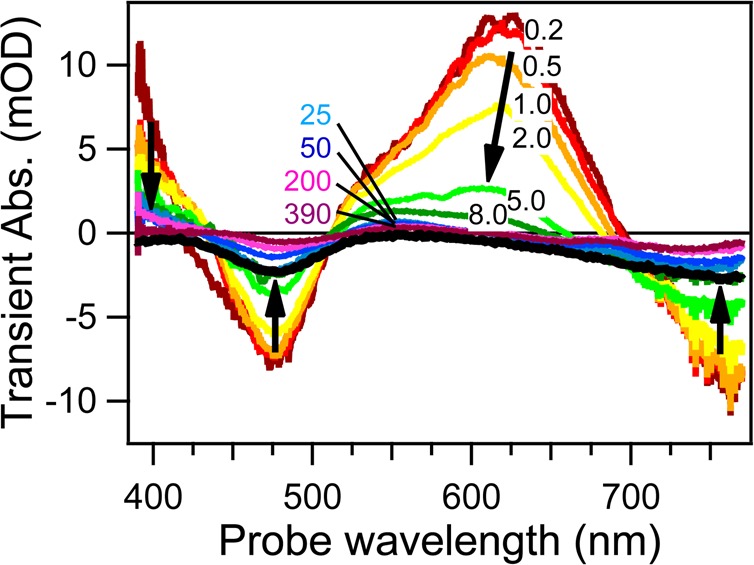
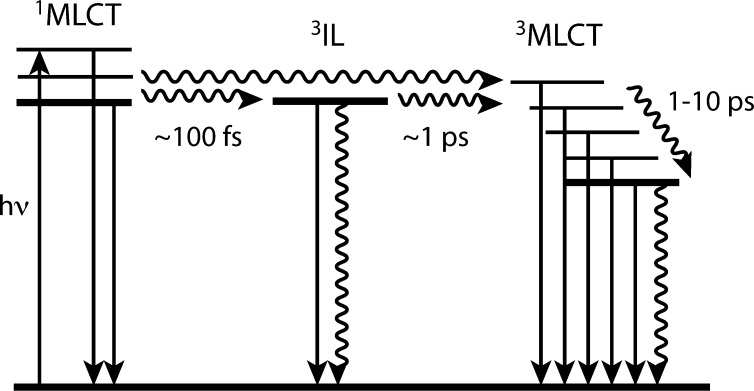
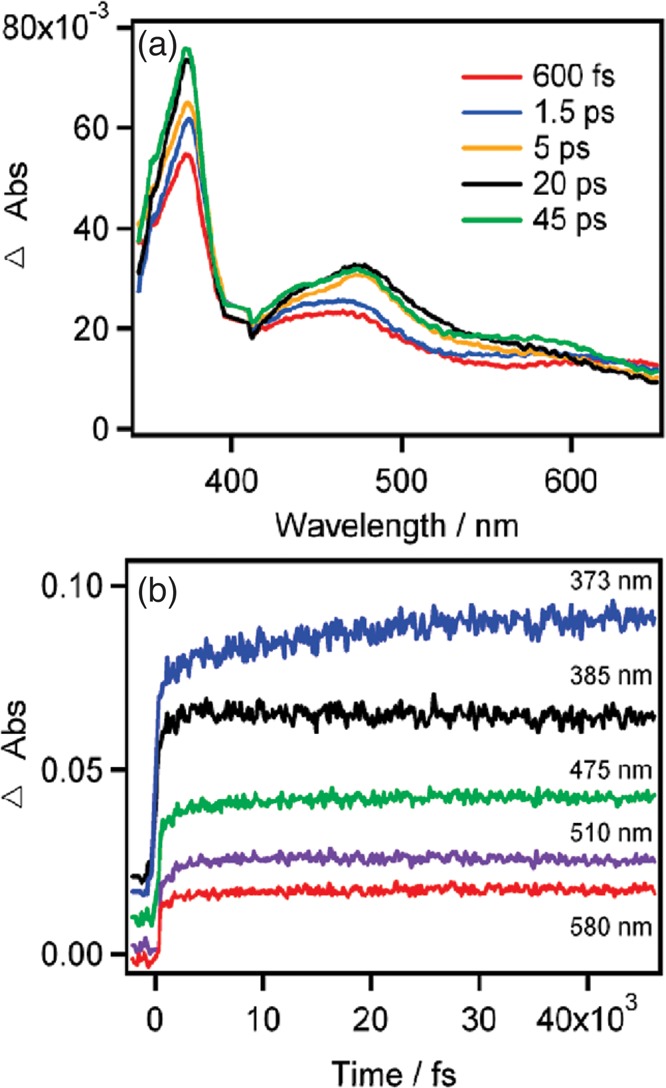
Rondi Ariana, Rodriguez Yuseff, Feurer Thomas, Cannizzo Andrea
Institute of Applied Physics, University of Bern, Sidlerstrasse 5, CH-3012 Bern, Switzerland.
Acc Chem Res. 2015 May 19;48(5):1432-40. doi: 10.1021/ar5003939. Epub 2015 Apr 22.
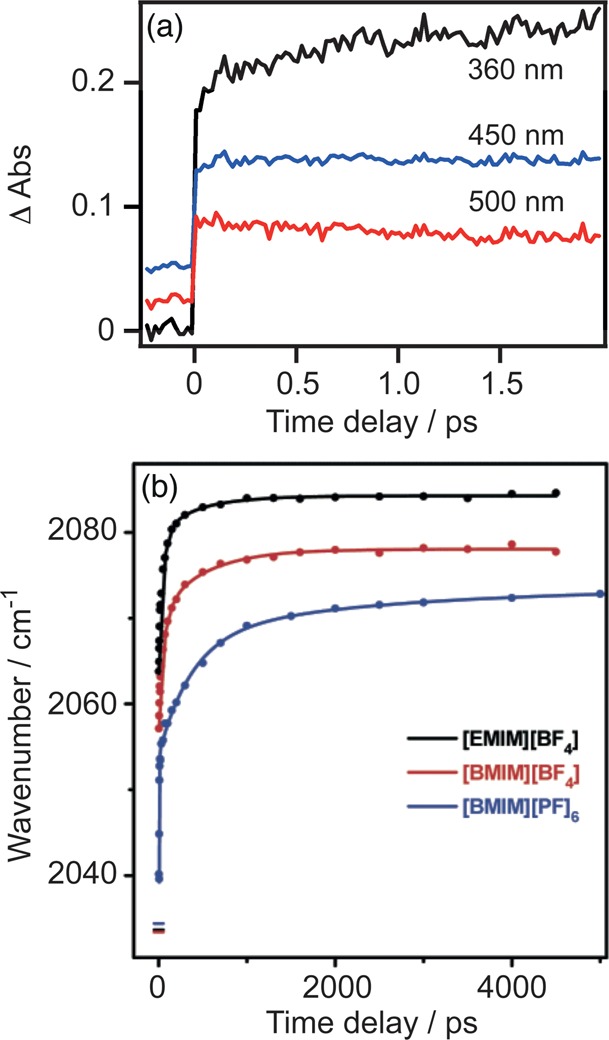
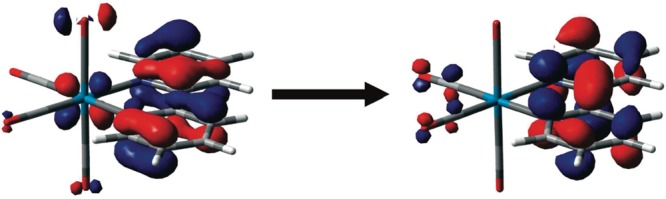
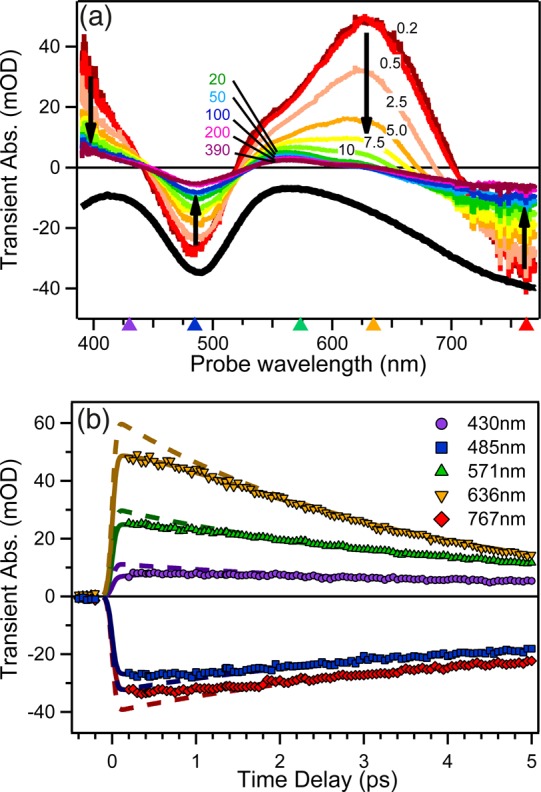
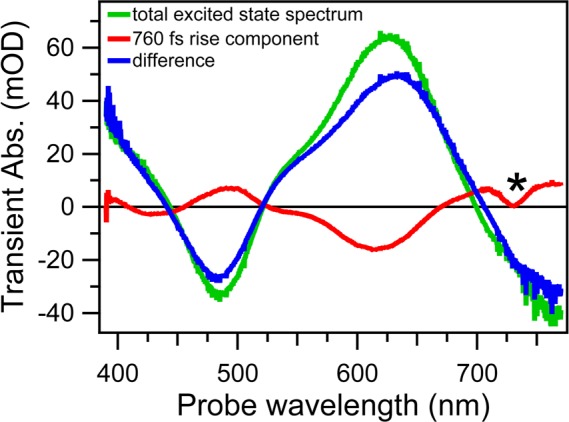
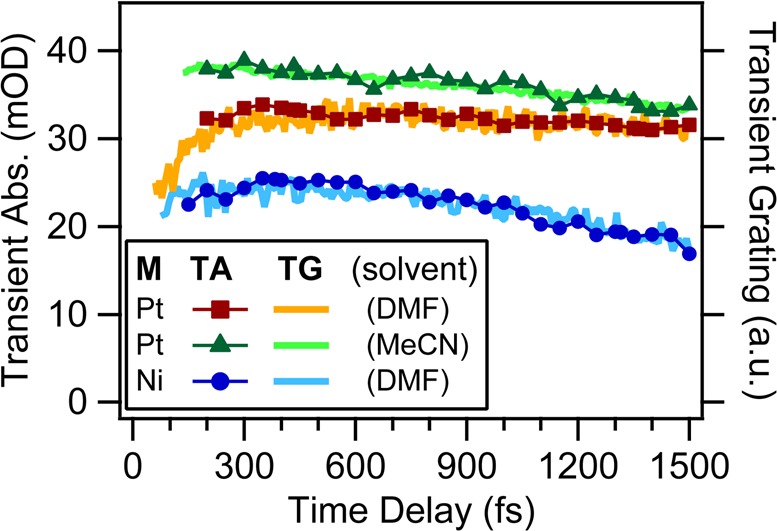
In any physicochemical process in liquids, the dynamical response of the solvent to the solutes out of equilibrium plays a crucial role in the rates and products: the solvent molecules react to the changes in volume and electron density of the solutes to minimize the free energy of the solution, thus modulating the activation barriers and stabilizing (or destabilizing) intermediate states. In charge transfer (CT) processes in polar solvents, the response of the solvent always assists the formation of charge separation states by stabilizing the energy of the localized charges. A deep understanding of the solvation mechanisms and time scales is therefore essential for a correct description of any photochemical process in dense phase and for designing molecular devices based on photosensitizers with CT excited states. In the last two decades, with the advent of ultrafast time-resolved spectroscopies, microscopic models describing the relevant case of polar solvation (where both the solvent and the solute molecules have a permanent electric dipole and the mutual interaction is mainly dipole-dipole) have dramatically progressed. Regardless of the details of each model, they all assume that the effect of the electrostatic fields of the solvent molecules on the internal electronic dynamics of the solute are perturbative and that the solvent-solute coupling is mainly an electrostatic interaction between the constant permanent dipoles of the solute and the solvent molecules. This well-established picture has proven to quantitatively rationalize spectroscopic effects of environmental and electric dynamics (time-resolved Stokes shifts, inhomogeneous broadening, etc.). However, recent computational and experimental studies, including ours, have shown that further improvement is required. Indeed, in the last years we investigated several molecular complexes exhibiting photoexcited CT states, and we found that the current description of the formation and stabilization of CT states in an important group of molecules such as transition metal complexes is inaccurate. In particular, we proved that the solvent molecules are not just spectators of intramolecular electron density redistribution but significantly modulate it. Our results solicit further development of quantum mechanics computational methods to treat the solute and (at least) the closest solvent molecules including the nonperturbative treatment of the effects of local electrostatics and direct solvent-solute interactions to describe the dynamical changes of the solute excited states during the solvent response.
在液体中的任何物理化学过程中,溶剂对处于非平衡态溶质的动力学响应在反应速率和产物方面起着关键作用:溶剂分子对溶质的体积和电子密度变化做出反应,以使溶液的自由能最小化,从而调节活化能垒并稳定(或不稳定)中间态。在极性溶剂中的电荷转移(CT)过程中,溶剂的响应总是通过稳定局域电荷的能量来促进电荷分离态的形成。因此,深入理解溶剂化机制和时间尺度对于正确描述密相中的任何光化学过程以及基于具有CT激发态的光敏剂设计分子器件至关重要。在过去的二十年中,随着超快时间分辨光谱学的出现,描述极性溶剂化相关情况(溶剂和溶质分子都具有永久电偶极且相互作用主要是偶极 - 偶极相互作用)的微观模型有了显著进展。无论每个模型的细节如何,它们都假定溶剂分子的静电场对溶质内部电子动力学的影响是微扰性的,并且溶剂 - 溶质耦合主要是溶质和溶剂分子的恒定永久偶极之间的静电相互作用。这一已确立的图景已被证明能够定量地解释环境和电动力学的光谱效应(时间分辨斯托克斯位移、非均匀展宽等)。然而,包括我们的研究在内,最近的计算和实验研究表明仍需要进一步改进。事实上,在过去几年中,我们研究了几种表现出光激发CT态的分子复合物,并且我们发现,对于诸如过渡金属配合物等重要分子组中CT态的形成和稳定的当前描述是不准确的。特别是,我们证明了溶剂分子不仅仅是分子内电子密度重新分布的旁观者,而是对其有显著的调节作用。我们的结果促使量子力学计算方法进一步发展,以处理溶质和(至少)最接近的溶剂分子,包括对局部静电效应和直接溶剂 - 溶质相互作用进行非微扰处理,以描述溶剂响应过程中溶质激发态的动态变化。