Kaplow Irene M, MacIsaac Julia L, Mah Sarah M, McEwen Lisa M, Kobor Michael S, Fraser Hunter B
Department of Computer Science, Stanford University, Stanford, California 94305, USA; Department of Biology, Stanford University, Stanford, California 94305, USA;
Centre for Molecular Medicine and Therapeutics, Child and Family Research Institute, University of British Columbia, Vancouver, British Columbia V5Z 4H4, Canada;
Genome Res. 2015 Jun;25(6):907-17. doi: 10.1101/gr.183749.114. Epub 2015 Apr 24.
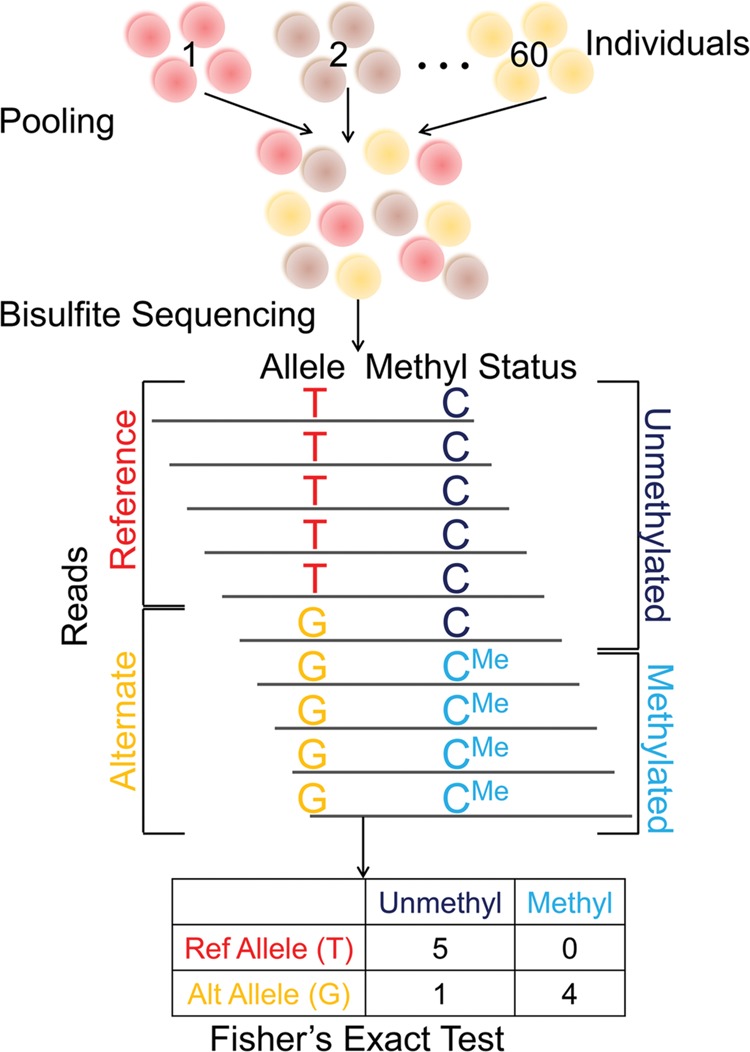
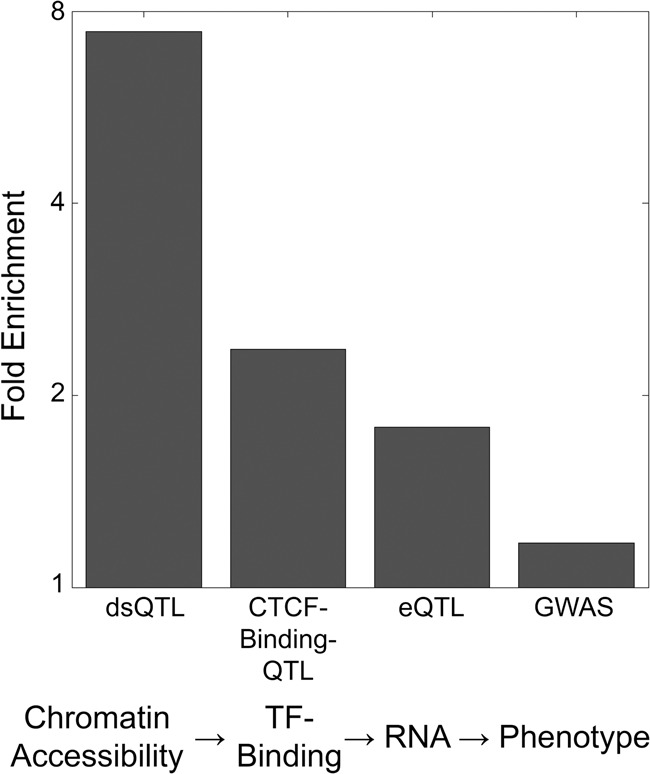
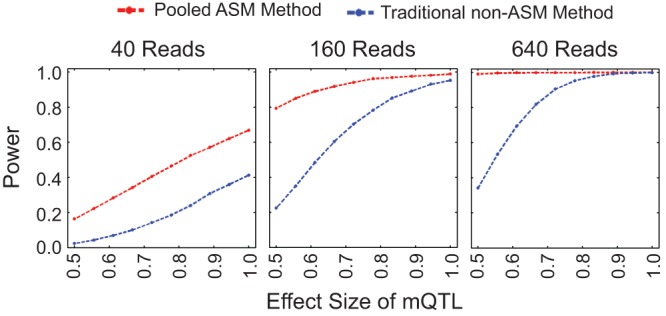
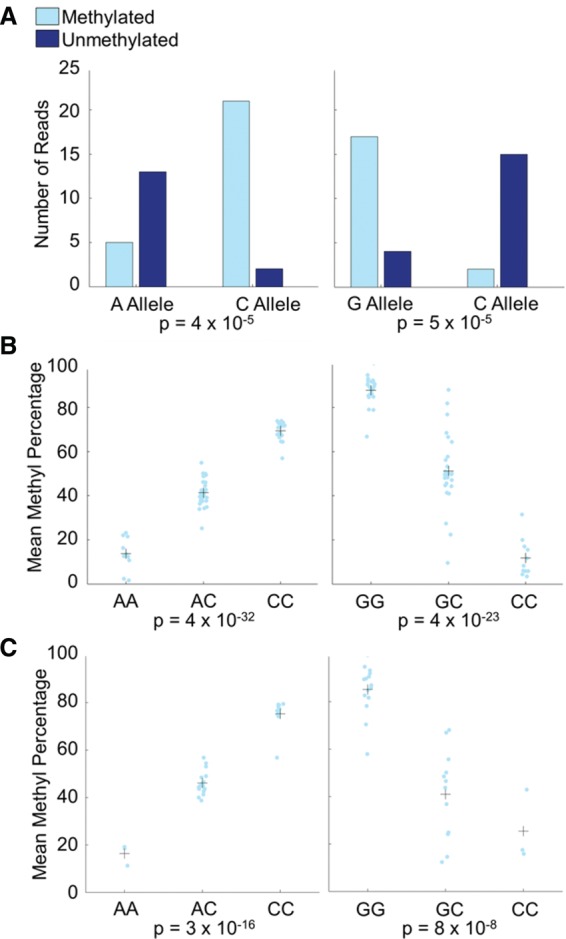
DNA methylation is an epigenetic modification that plays a key role in gene regulation. Previous studies have investigated its genetic basis by mapping genetic variants that are associated with DNA methylation at specific sites, but these have been limited to microarrays that cover <2% of the genome and cannot account for allele-specific methylation (ASM). Other studies have performed whole-genome bisulfite sequencing on a few individuals, but these lack statistical power to identify variants associated with DNA methylation. We present a novel approach in which bisulfite-treated DNA from many individuals is sequenced together in a single pool, resulting in a truly genome-wide map of DNA methylation. Compared to methods that do not account for ASM, our approach increases statistical power to detect associations while sharply reducing cost, effort, and experimental variability. As a proof of concept, we generated deep sequencing data from a pool of 60 human cell lines; we evaluated almost twice as many CpGs as the largest microarray studies and identified more than 2000 genetic variants associated with DNA methylation. We found that these variants are highly enriched for associations with chromatin accessibility and CTCF binding but are less likely to be associated with traits indirectly linked to DNA, such as gene expression and disease phenotypes. In summary, our approach allows genome-wide mapping of genetic variants associated with DNA methylation in any tissue of any species, without the need for individual-level genotype or methylation data.
DNA甲基化是一种表观遗传修饰,在基因调控中起关键作用。以往的研究通过绘制与特定位点DNA甲基化相关的遗传变异来探究其遗传基础,但这些研究仅限于覆盖不到2%基因组的微阵列,且无法解释等位基因特异性甲基化(ASM)。其他研究对少数个体进行了全基因组亚硫酸氢盐测序,但这些研究缺乏识别与DNA甲基化相关变异的统计能力。我们提出了一种新方法,将来自许多个体的亚硫酸氢盐处理后的DNA在一个池中一起测序,从而得到一个真正全基因组范围的DNA甲基化图谱。与不考虑ASM的方法相比,我们的方法在大幅降低成本、工作量和实验变异性的同时,提高了检测关联的统计能力。作为概念验证,我们从60个人类细胞系的一个池中生成了深度测序数据;我们评估的CpG数量几乎是最大规模微阵列研究的两倍,并鉴定出2000多个与DNA甲基化相关的遗传变异。我们发现,这些变异与染色质可及性和CTCF结合的关联高度富集,但与间接与DNA相关的性状(如基因表达和疾病表型)的关联可能性较小。总之,我们的方法允许在任何物种的任何组织中对与DNA甲基化相关的遗传变异进行全基因组图谱绘制,而无需个体水平的基因型或甲基化数据。