Netravathi Manjunath, Kumari Renu, Kapoor Saketh, Dakle Pushkar, Dwivedi Manish Kumar, Roy Sumitabho Deb, Pandey Paritosh, Saini Jitender, Ramakrishna Anil, Navalli Devaraddi, Satishchandra Parthasarathy, Pal Pramod Kumar, Kumar Arun, Faruq Mohammed
Department of Neurology, National Institute of mental health & Neurosciences (NIMHANS), Bangalore, 560029, India.
Genomics and Molecular Medicine, CSIR-Institute of Genomics and Integrative Biology, Mall Road, New Delhi, India.
BMC Med Genet. 2015 Feb 10;16:5. doi: 10.1186/s12881-015-0151-8.
Coats plus syndrome is an autosomal recessive, pleiotropic, multisystem disorder characterized by retinal telangiectasia and exudates, intracranial calcification with leukoencephalopathy and brain cysts, osteopenia with predisposition to fractures, bone marrow suppression, gastrointestinal bleeding and portal hypertension. It is caused by compound heterozygous mutations in the CTC1 gene.
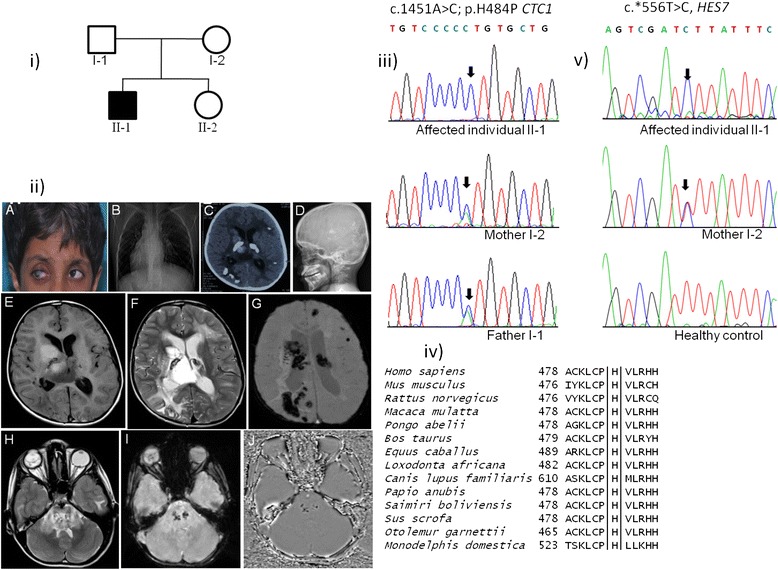
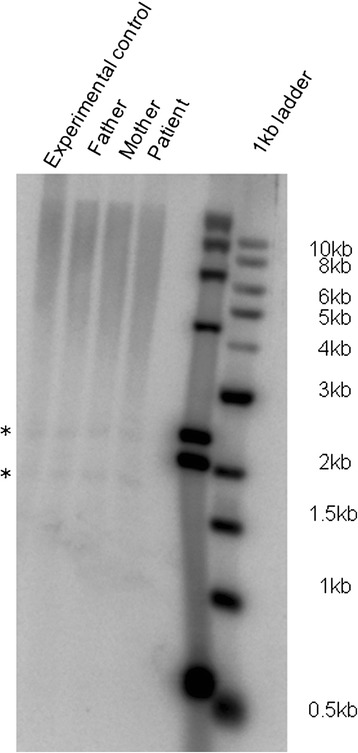
We encountered a case of an eight-year old boy from an Indian family with manifestations of Coats plus syndrome along with an unusual occurrence of dextrocardia and situs inversus. Targeted resequencing of the CTC1 gene as well as whole exome sequencing (WES) were conducted in this family to identify the causal variations. The identified candidate variations were screened in ethnicity matched healthy controls. The effect of CTC1 variation on telomere length was assessed using Southern blot. A novel homozygous missense mutation c.1451A > C (p.H484P) in exon 9 of the CTC1 gene and a rare 3'UTR known dbSNP variation (c.*556 T > C) in HES7 were identified as the plausible candidates associated with this complex phenotype of Coats plus and dextrocardia. This CTC1 variation was absent in the controls and we also observed a reduced telomere length in the affected individual's DNA, suggesting its likely pathogenic nature. The reported p.H484P mutation is located in the N-terminal 700 amino acid regionthat is important for the binding of CTC1 to ssDNA through its two OB domains. WES data also showed a rare homozygous missense variation in the TEK gene in the affected individual. Both HES7 and TEK are targets of the Notch signaling pathway.
This is the first report of a genetically confirmed case of Coats plus syndrome from India. By means of WES, the genetic variations in this family with unique and rare complex phenotype could be traced effectively. We speculate the important role of Notch signaling in this complex phenotypic presentation of Coats plus syndrome and dextrocardia. The present finding will be useful for genetic diagnosis and carrier detection in the family and for other patients with similar disease manifestations.
科茨加综合征是一种常染色体隐性、多效性、多系统疾病,其特征为视网膜毛细血管扩张和渗出、伴有白质脑病和脑囊肿的颅内钙化、易发生骨折的骨质减少、骨髓抑制、胃肠道出血和门静脉高压。它由CTC1基因的复合杂合突变引起。
我们遇到一例来自印度家庭的8岁男孩,患有科茨加综合征,并伴有罕见的右位心和内脏反位现象。对该家庭进行了CTC1基因的靶向重测序以及全外显子组测序(WES),以确定致病变异。在种族匹配的健康对照中筛选已鉴定的候选变异。使用Southern印迹法评估CTC1变异对端粒长度的影响。在CTC1基因第9外显子中鉴定出一个新的纯合错义突变c.1451A>C(p.H484P),以及在HES7中一个罕见的3'UTR已知dbSNP变异(c.*556 T>C),被确定为与科茨加综合征和右位心这种复杂表型相关的可能候选变异。该CTC1变异在对照中不存在,并且我们还观察到患病个体的DNA中端粒长度缩短,表明其可能具有致病性。报道的p.H484P突变位于N端700个氨基酸区域,该区域对于CTC1通过其两个OB结构域与单链DNA结合很重要。WES数据还显示患病个体的TEK基因存在罕见的纯合错义变异。HES7和TEK都是Notch信号通路的靶点。
这是印度首例经基因确诊的科茨加综合征病例报告。通过WES,能够有效追踪这个具有独特罕见复杂表型家庭中的基因变异。我们推测Notch信号在科茨加综合征和右位心这种复杂表型表现中起重要作用。目前的发现将有助于该家庭的基因诊断和携带者检测,以及其他有类似疾病表现的患者。