Yamasaki Hiroyuki, Funai Yusuke, Funao Tomoharu, Mori Takashi, Nishikawa Kiyonobu
Department of Anesthesiology, Osaka City University Graduate School of Medicine, Osaka, Japan.
PLoS One. 2015 May 1;10(5):e0125147. doi: 10.1371/journal.pone.0125147. eCollection 2015.
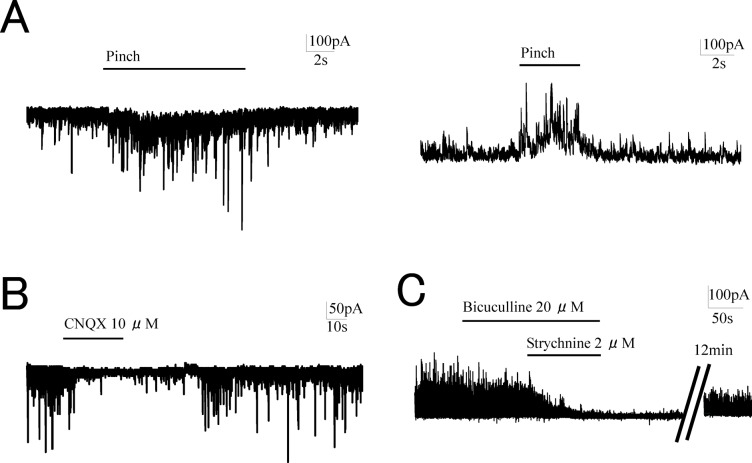
Tramadol is thought to modulate synaptic transmissions in the spinal dorsal horn mainly by activating µ-opioid receptors and by inhibiting the reuptake of monoamines in the CNS. However, the precise mode of modulation remains unclear. We used an in vivo patch clamp technique in urethane-anesthetized rats to determine the antinociceptive mechanism of tramadol. In vivo whole-cell recordings of spontaneous inhibitory postsynaptic currents (sIPSCs) and spontaneous excitatory postsynaptic currents (sEPSCs) were made from substantia gelatinosa (SG) neurons (lamina II) at holding potentials of 0 mV and -70 mV, respectively. The effects of intravenous administration (0.5, 5, 15 mg/kg) of tramadol were evaluated. The effects of superfusion of tramadol on the surface of the spinal cord and of a tramadol metabolite (M1) were further analyzed. Intravenous administration of tramadol at doses >5 mg/kg decreased the sEPSCs and increased the sIPSCs in SG neurons. These effects were not observed following naloxone pretreatment. Tramadol superfusion at a clinically relevant concentration (10 µM) had no effect, but when administered at a very high concentration (100 µM), tramadol decreased sEPSCs, produced outward currents, and enhanced sIPSCs. The effects of M1 (1, 5 mg/kg intravenously) on sEPSCs and sIPSCs were similar to those of tramadol at a corresponding dose (5, 15 mg/kg). The present study demonstrated that systemically administered tramadol indirectly inhibited glutamatergic transmission, and enhanced GABAergic and glycinergic transmissions in SG neurons. These effects were mediated primarily by the activation of μ-opioid receptors. M1 may play a key role in the antinociceptive mechanisms of tramadol.
曲马多被认为主要通过激活μ-阿片受体和抑制中枢神经系统中单胺的再摄取来调节脊髓背角的突触传递。然而,确切的调节方式仍不清楚。我们在乌拉坦麻醉的大鼠中使用体内膜片钳技术来确定曲马多的抗伤害感受机制。分别在0 mV和 -70 mV的钳制电位下,从脊髓胶状质(SG)神经元(II层)进行体内全细胞记录自发抑制性突触后电流(sIPSCs)和自发兴奋性突触后电流(sEPSCs)。评估了静脉注射(0.5、5、15 mg/kg)曲马多的效果。进一步分析了曲马多在脊髓表面的灌流作用以及曲马多代谢物(M1)的作用。静脉注射剂量>5 mg/kg的曲马多可降低SG神经元中的sEPSCs并增加sIPSCs。纳洛酮预处理后未观察到这些效应。临床相关浓度(10 μM)的曲马多灌流无作用,但以非常高的浓度(100 μM)给药时,曲马多可降低sEPSCs,产生外向电流,并增强sIPSCs。M1(静脉注射1、5 mg/kg)对sEPSCs和sIPSCs的作用与相应剂量(5、15 mg/kg)的曲马多相似。本研究表明,全身给药的曲马多间接抑制SG神经元中的谷氨酸能传递,并增强GABA能和甘氨酸能传递。这些效应主要由μ-阿片受体的激活介导。M1可能在曲马多的抗伤害感受机制中起关键作用。