Alfonso-Morales Abdulahi, Rios Liliam, Martínez-Pérez Orlando, Dolz Roser, Valle Rosa, Perera Carmen L, Bertran Kateri, Frías Maria T, Ganges Llilianne, Díaz de Arce Heidy, Majó Natàlia, Núñez José I, Pérez Lester J
Centro Nacional de Sanidad Agropecuaria (CENSA), La Habana, Cuba.
Universidad de las Ciencias Informáticas (UCI), La Habana, Cuba.
PLoS One. 2015 May 6;10(5):e0125853. doi: 10.1371/journal.pone.0125853. eCollection 2015.
Infectious bursal disease (IBD) is a highly contagious and acute viral disease, which has caused high mortality rates in birds and considerable economic losses in different parts of the world for more than two decades and it still represents a considerable threat to poultry. The current study was designed to rigorously measure the reliability of a phylogenetic marker included into segment B. This marker can facilitate molecular epidemiology studies, incorporating this segment of the viral genome, to better explain the links between emergence, spreading and maintenance of the very virulent IBD virus (vvIBDV) strains worldwide.
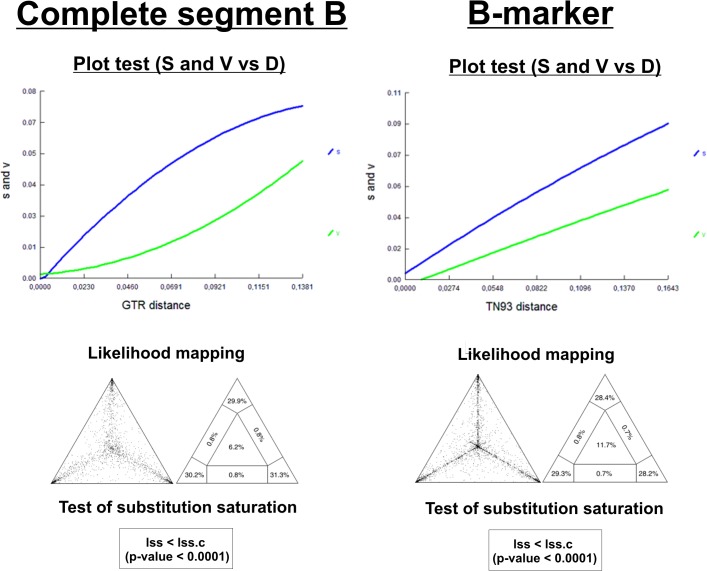
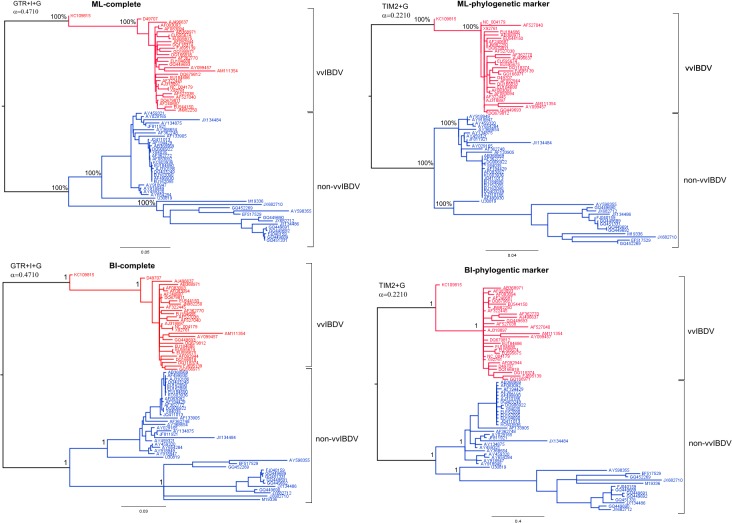
METHODOLOGY/PRINCIPAL FINDINGS: Sequences of the segment B gene from IBDV strains isolated from diverse geographic locations were obtained from the GenBank Database; Cuban sequences were obtained in the current work. A phylogenetic marker named B-marker was assessed by different phylogenetic principles such as saturation of substitution, phylogenetic noise and high consistency. This last parameter is based on the ability of B-marker to reconstruct the same topology as the complete segment B of the viral genome. From the results obtained from B-marker, demographic history for both main lineages of IBDV regarding segment B was performed by Bayesian skyline plot analysis. Phylogenetic analysis for both segments of IBDV genome was also performed, revealing the presence of a natural reassortant strain with segment A from vvIBDV strains and segment B from non-vvIBDV strains within Cuban IBDV population.
CONCLUSIONS/SIGNIFICANCE: This study contributes to a better understanding of the emergence of vvIBDV strains, describing molecular epidemiology of IBDV using the state-of-the-art methodology concerning phylogenetic reconstruction. This study also revealed the presence of a novel natural reassorted strain as possible manifest of change in the genetic structure and stability of the vvIBDV strains. Therefore, it highlights the need to obtain information about both genome segments of IBDV for molecular epidemiology studies.
传染性法氏囊病(IBD)是一种高度传染性的急性病毒性疾病,二十多年来在世界各地导致禽类高死亡率和巨大经济损失,至今仍对家禽构成重大威胁。本研究旨在严格测定B节段中一个系统发育标记的可靠性。该标记有助于分子流行病学研究,纳入病毒基因组的这一片段,以更好地解释超强毒传染性法氏囊病病毒(vvIBDV)毒株在全球出现、传播和维持之间的联系。
方法/主要发现:从GenBank数据库获得了来自不同地理位置的传染性法氏囊病病毒毒株B节段基因的序列;古巴的序列是在当前研究中获得的。一个名为B标记的系统发育标记通过不同的系统发育原则进行评估,如替换饱和度、系统发育噪声和高一致性。最后一个参数基于B标记重建与病毒基因组完整B节段相同拓扑结构的能力。根据B标记获得的结果,通过贝叶斯天际线图分析对传染性法氏囊病病毒两个主要谱系关于B节段的种群历史进行了研究。还对传染性法氏囊病病毒基因组的两个节段进行了系统发育分析,揭示了古巴传染性法氏囊病病毒种群中存在一种天然重配毒株,其A节段来自vvIBDV毒株,B节段来自非vvIBDV毒株。
结论/意义:本研究有助于更好地理解vvIBDV毒株的出现,使用关于系统发育重建的最新方法描述传染性法氏囊病病毒的分子流行病学。本研究还揭示了一种新型天然重配毒株的存在,这可能是vvIBDV毒株遗传结构和稳定性变化的表现。因此,它强调了在分子流行病学研究中获取传染性法氏囊病病毒两个基因组节段信息的必要性。