Smith Rob, Taylor Ryan M, Prince John T
BMC Bioinformatics. 2015;16 Suppl 7(Suppl 7):S2. doi: 10.1186/1471-2105-16-S7-S2. Epub 2015 Apr 23.
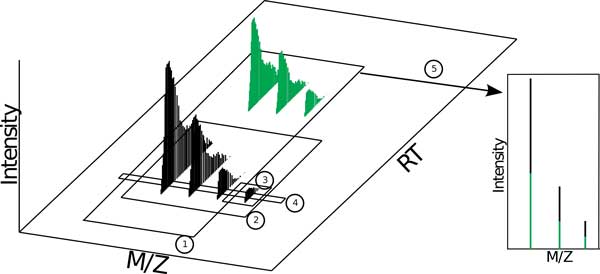
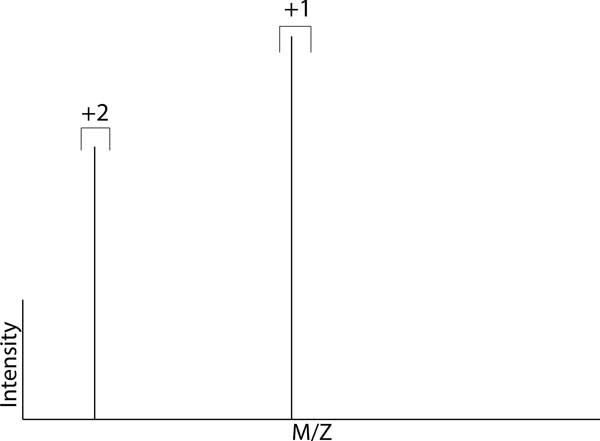
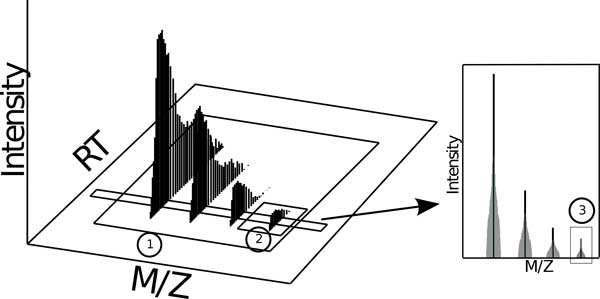
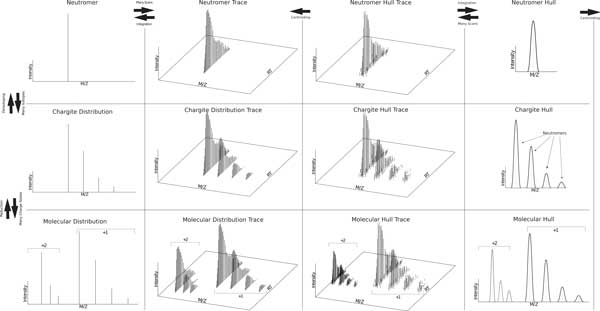
The comparison of analyte mass spectrometry precursor (MS1) signal is central to many proteomic (and other -omic) workflows. Standard vocabularies for mass spectrometry exist and provide good coverage for most experimental applications yet are insufficient for concise and unambiguous description of data concepts spanning the range of signal provenance from a molecular perspective (e.g. from charged peptides down to fine isotopes). Without a standard unambiguous nomenclature, literature searches, algorithm reproducibility and algorithm evaluation for MS-omics data processing are nearly impossible.
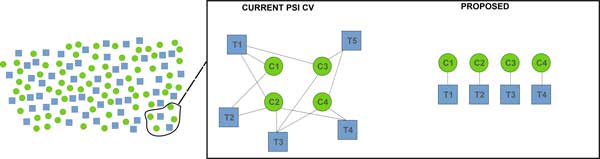
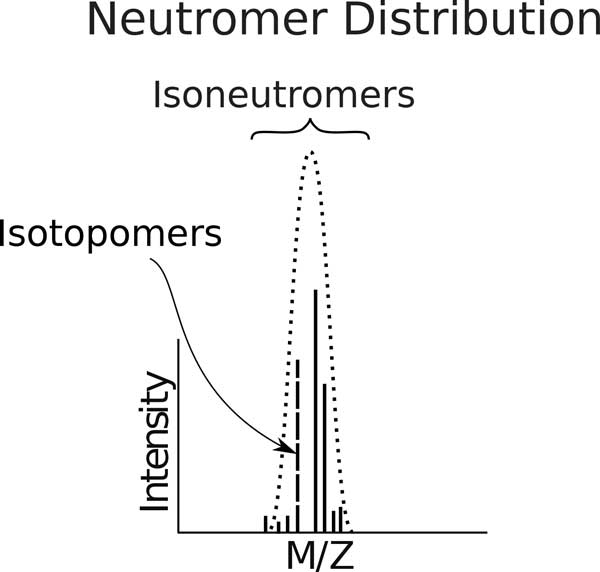
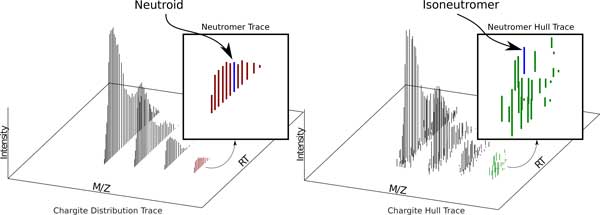
We show how terms from current official ontologies are too vague or ambiguous to explicitly map molecular entities to MS signals and we illustrate the inconsistency and ambiguity of current colloquially used terms. We also propose a set of terms for MS1 signal that uniquely, succinctly and intuitively describe data concepts spanning the range of signal provenance from full molecule downs to fine isotopes. We suggest that additional community discussion of these terms should precede any further standardization efforts. We propose a novel nomenclature that spans the range of the required granularity to describe MS data processing from the perspective of the molecular provenance of the MS signal.
The proposed nomenclature provides a chain of succinct and unique terms spanning the signal created by a charged molecule down through each of its constituent subsignals. We suggest that additional community discussion of these terms should precede any further standardization efforts.
分析物质谱前体(MS1)信号的比较是许多蛋白质组学(以及其他组学)工作流程的核心。质谱的标准词汇表已经存在,并且对大多数实验应用提供了良好的覆盖,但从分子角度(例如从带电肽到精细同位素)对跨越信号来源范围的数据概念进行简洁明了且无歧义的描述还不够。没有标准的无歧义命名法,对质谱组学数据处理进行文献检索、算法重现性和算法评估几乎是不可能的。
我们展示了当前官方本体中的术语如何过于模糊或有歧义,以至于无法将分子实体明确映射到质谱信号,并且我们说明了当前常用术语的不一致性和歧义性。我们还提出了一组用于MS1信号的术语,这些术语能够独特、简洁且直观地描述从完整分子到精细同位素的信号来源范围内的数据概念。我们建议在进行任何进一步的标准化工作之前,应先对这些术语进行更多的社区讨论。我们提出了一种新颖的命名法,其涵盖了所需粒度范围,以便从质谱信号的分子来源角度描述质谱数据处理。
所提出的命名法提供了一系列简洁且独特的术语,涵盖了由带电分子产生的信号及其每个组成子信号。我们建议在进行任何进一步的标准化工作之前,应先对这些术语进行更多的社区讨论。