Wetmore Kelly M, Price Morgan N, Waters Robert J, Lamson Jacob S, He Jennifer, Hoover Cindi A, Blow Matthew J, Bristow James, Butland Gareth, Arkin Adam P, Deutschbauer Adam
Physical Biosciences Division, Lawrence Berkeley National Laboratory, Berkeley, California, USA.
Joint Genome Institute, Lawrence Berkeley National Laboratory, Walnut Creek, California, USA.
mBio. 2015 May 12;6(3):e00306-15. doi: 10.1128/mBio.00306-15.
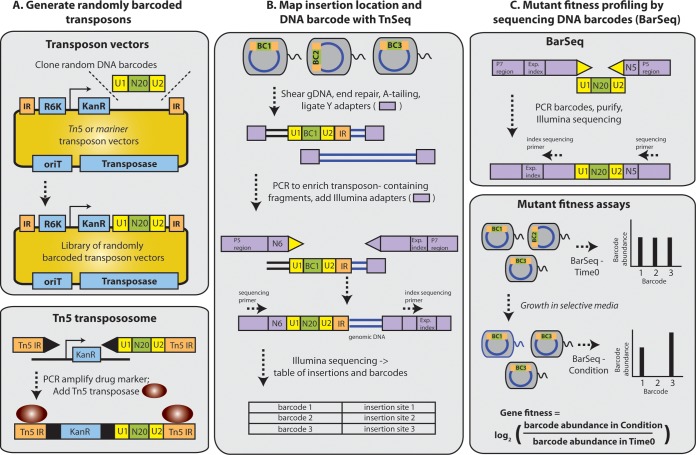
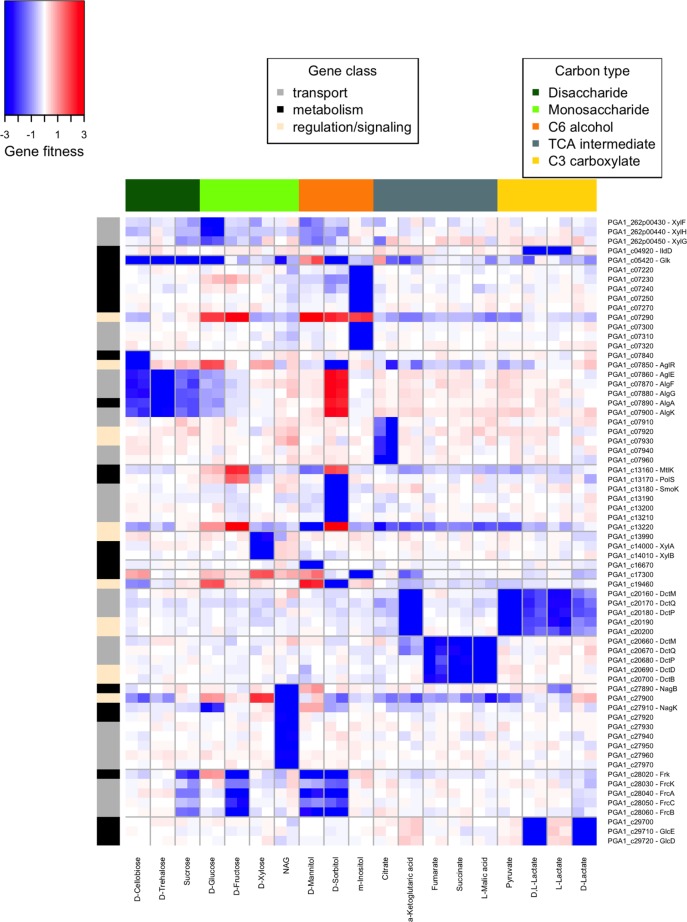
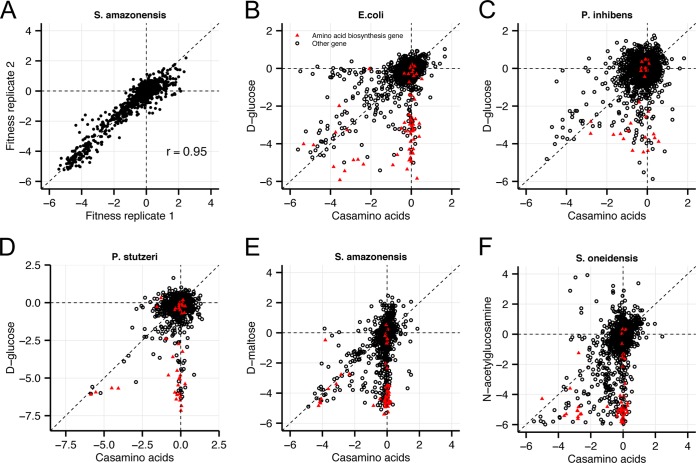
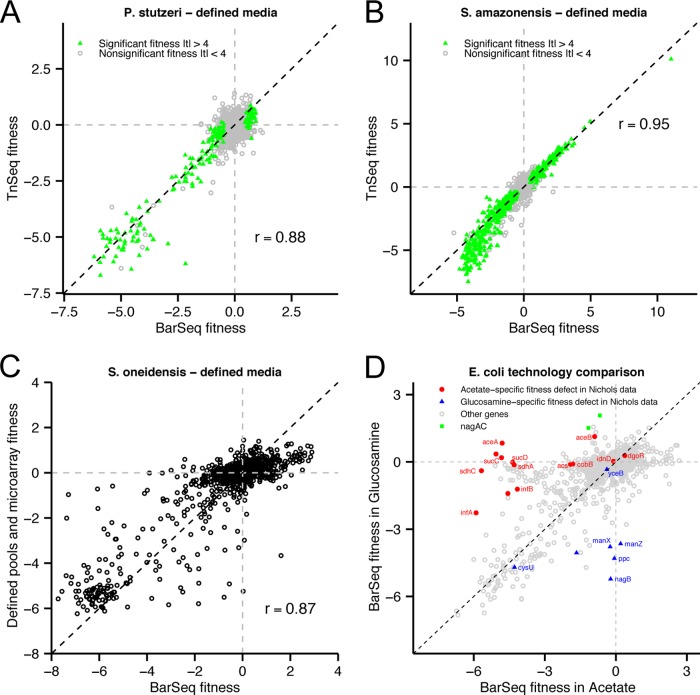
Transposon mutagenesis with next-generation sequencing (TnSeq) is a powerful approach to annotate gene function in bacteria, but existing protocols for TnSeq require laborious preparation of every sample before sequencing. Thus, the existing protocols are not amenable to the throughput necessary to identify phenotypes and functions for the majority of genes in diverse bacteria. Here, we present a method, random bar code transposon-site sequencing (RB-TnSeq), which increases the throughput of mutant fitness profiling by incorporating random DNA bar codes into Tn5 and mariner transposons and by using bar code sequencing (BarSeq) to assay mutant fitness. RB-TnSeq can be used with any transposon, and TnSeq is performed once per organism instead of once per sample. Each BarSeq assay requires only a simple PCR, and 48 to 96 samples can be sequenced on one lane of an Illumina HiSeq system. We demonstrate the reproducibility and biological significance of RB-TnSeq with Escherichia coli, Phaeobacter inhibens, Pseudomonas stutzeri, Shewanella amazonensis, and Shewanella oneidensis. To demonstrate the increased throughput of RB-TnSeq, we performed 387 successful genome-wide mutant fitness assays representing 130 different bacterium-carbon source combinations and identified 5,196 genes with significant phenotypes across the five bacteria. In P. inhibens, we used our mutant fitness data to identify genes important for the utilization of diverse carbon substrates, including a putative d-mannose isomerase that is required for mannitol catabolism. RB-TnSeq will enable the cost-effective functional annotation of diverse bacteria using mutant fitness profiling.
A large challenge in microbiology is the functional assessment of the millions of uncharacterized genes identified by genome sequencing. Transposon mutagenesis coupled to next-generation sequencing (TnSeq) is a powerful approach to assign phenotypes and functions to genes. However, the current strategies for TnSeq are too laborious to be applied to hundreds of experimental conditions across multiple bacteria. Here, we describe an approach, random bar code transposon-site sequencing (RB-TnSeq), which greatly simplifies the measurement of gene fitness by using bar code sequencing (BarSeq) to monitor the abundance of mutants. We performed 387 genome-wide fitness assays across five bacteria and identified phenotypes for over 5,000 genes. RB-TnSeq can be applied to diverse bacteria and is a powerful tool to annotate uncharacterized genes using phenotype data.
利用下一代测序技术进行转座子诱变(TnSeq)是一种注释细菌基因功能的强大方法,但现有的TnSeq方案在测序前需要对每个样本进行繁琐的制备。因此,现有方案无法满足鉴定多种细菌中大多数基因的表型和功能所需的通量。在此,我们提出一种方法——随机条形码转座子位点测序(RB-TnSeq),通过将随机DNA条形码整合到Tn5和水手座转座子中,并使用条形码测序(BarSeq)来测定突变体适应性,从而提高突变体适应性分析的通量。RB-TnSeq可与任何转座子一起使用,并且每个生物体只需进行一次TnSeq,而不是每个样本进行一次。每次BarSeq分析仅需简单的PCR,并且在Illumina HiSeq系统的一个泳道上可对48至96个样本进行测序。我们用大肠杆菌、抑制发光杆菌、施氏假单胞菌、亚马逊希瓦氏菌和单胞希瓦氏菌证明了RB-TnSeq的可重复性和生物学意义。为了证明RB-TnSeq通量的提高,我们进行了387次成功的全基因组突变体适应性分析,涵盖130种不同的细菌-碳源组合,并在这五种细菌中鉴定出5196个具有显著表型的基因。在抑制发光杆菌中,我们利用突变体适应性数据鉴定了对多种碳底物利用重要的基因,包括一种参与甘露醇分解代谢所需的假定的D-甘露糖异构酶。RB-TnSeq将能够通过突变体适应性分析对多种细菌进行经济高效的功能注释。
微生物学中的一个重大挑战是对通过基因组测序鉴定出的数百万个未表征基因进行功能评估。转座子诱变与下一代测序技术相结合(TnSeq)是一种为基因赋予表型和功能的强大方法。然而,当前的TnSeq策略过于繁琐,无法应用于多种细菌的数百种实验条件。在此,我们描述了一种方法——随机条形码转座子位点测序(RB-TnSeq),它通过使用条形码测序(BarSeq)来监测突变体丰度,极大地简化了基因适应性的测量。我们对五种细菌进行了387次全基因组适应性分析,并鉴定出5000多个基因的表型。RB-TnSeq可应用于多种细菌,是一种利用表型数据注释未表征基因的强大工具。