Siarkou Victoria I, Vorimore Fabien, Vicari Nadia, Magnino Simone, Rodolakis Annie, Pannekoek Yvonne, Sachse Konrad, Longbottom David, Laroucau Karine
Laboratory of Microbiology and Infectious Diseases, School of Veterinary Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki, Thessaloniki, Greece.
Anses, Animal Health Laboratory, Bacterial Zoonoses Unit, University Paris-Est, Maisons-Alfort, France.
PLoS One. 2015 May 22;10(5):e0126433. doi: 10.1371/journal.pone.0126433. eCollection 2015.
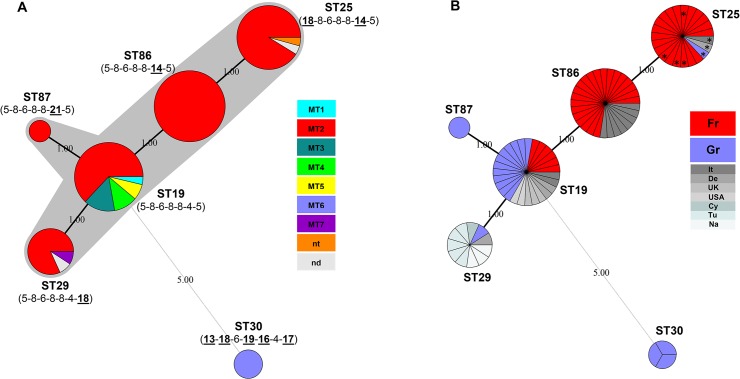
Chlamydia abortus, an obligate intracellular bacterium, is the most common infectious cause of abortion in small ruminants worldwide and has zoonotic potential. We applied multilocus sequence typing (MLST) together with multiple-locus variable-number tandem repeat analysis (MLVA) to genotype 94 ruminant C. abortus strains, field isolates and samples collected from 1950 to 2011 in diverse geographic locations, with the aim of delineating C. abortus lineages and clones. MLST revealed the previously identified sequence types (STs) ST19, ST25, ST29 and ST30, plus ST86, a recently-assigned type on the Chlamydiales MLST website and ST87, a novel type harbouring the hemN_21 allele, whereas MLVA recognized seven types (MT1 to MT7). Minimum-spanning-tree analysis suggested that all STs but one (ST30) belonged to a single clonal complex, possibly reflecting the short evolutionary timescale over which the predicted ancestor (ST19) has diversified into three single-locus variants (ST86, ST87 and ST29) and further, through ST86 diversification, into one double-locus variant (ST25). ST descendants have probably arisen through a point mutation evolution mode. Interestingly, MLVA showed that in the ST19 population there was a greater genetic diversity than in other STs, most of which exhibited the same MT over time and geographical distribution. However, the evolutionary pathways of C. abortus STs seem to be diverse across geographic distances with individual STs restricted to particular geographic locations. The ST30 singleton clone displaying geographic specificity and represented by the Greek strains LLG and POS was effectively distinguished from the clonal complex lineage, supporting the notion that possibly two separate host adaptations and hence independent bottlenecks of C. abortus have occurred through time. The combination of MLST and MLVA assays provides an additional level of C. abortus discrimination and may prove useful for the investigation and surveillance of emergent C. abortus clonal populations.
流产衣原体是一种专性胞内细菌,是全球小型反刍动物流产最常见的感染原因,且具有人畜共患病潜力。我们应用多位点序列分型(MLST)以及多位点可变数目串联重复序列分析(MLVA)对94株反刍动物流产衣原体菌株、1950年至2011年期间在不同地理位置采集的现场分离株和样本进行基因分型,目的是描绘流产衣原体谱系和克隆。MLST揭示了先前鉴定的序列类型(STs)ST19、ST25、ST29和ST30,以及衣原体MLST网站上最近指定的ST86和携带hemN_21等位基因的新型ST87,而MLVA识别出七种类型(MT1至MT7)。最小生成树分析表明,除一种(ST30)外,所有STs都属于一个单一的克隆复合体,这可能反映了预测祖先(ST19)分化为三个单一位点变体(ST86、ST87和ST29)的进化时间尺度较短,并且通过ST86的多样化进一步分化为一个双位点变体(ST25)。ST后代可能是通过点突变进化模式产生的。有趣的是,MLVA表明,在ST19群体中,遗传多样性比其他STs更大,其中大多数在时间和地理分布上表现出相同的MT。然而,流产衣原体STs的进化途径似乎在不同地理距离上各不相同,个别STs局限于特定地理位置。由希腊菌株LLG和POS代表的具有地理特异性的ST30单克隆克隆有效地与克隆复合体谱系区分开来,支持了随着时间推移可能发生了两种独立的宿主适应性以及因此流产衣原体独立瓶颈的观点。MLST和MLVA检测的结合提供了流产衣原体鉴别水平的提升,可能对新兴流产衣原体克隆群体的调查和监测有用。