Haulcomb Melissa M, Mesnard-Hoaglin Nichole A, Batka Richard J, Meadows Rena M, Miller Whitney M, Mcmillan Kathryn P, Brown Todd J, Sanders Virginia M, Jones Kathryn J
Neuroscience Program, Loyola University Medical Center, Maywood, Illinois, 60153.
Research and Development Service, Hines Veterans Administration Hospital, Hines, Illinois, 60141.
J Comp Neurol. 2015 Dec 15;523(18):2752-68. doi: 10.1002/cne.23814. Epub 2015 Jun 22.
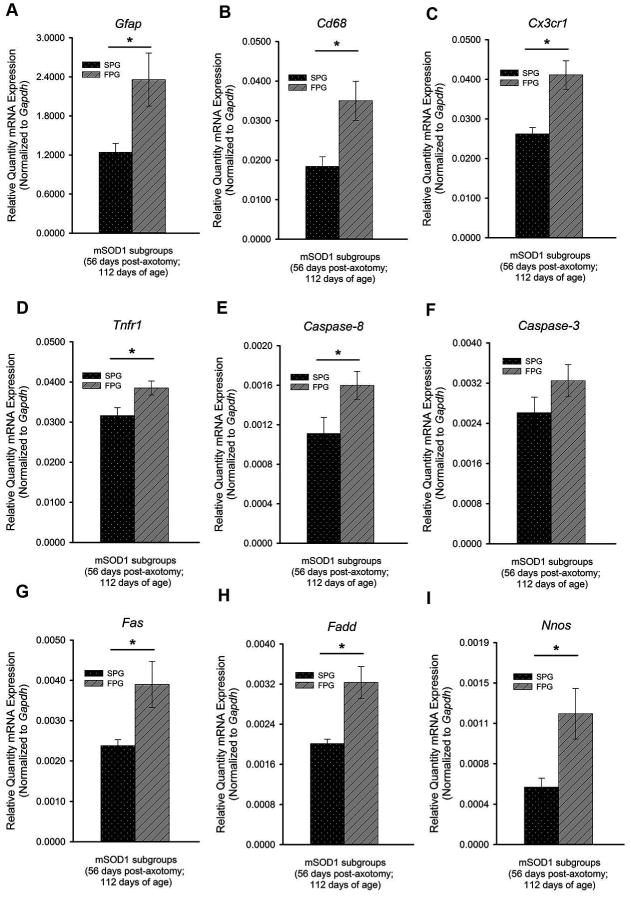
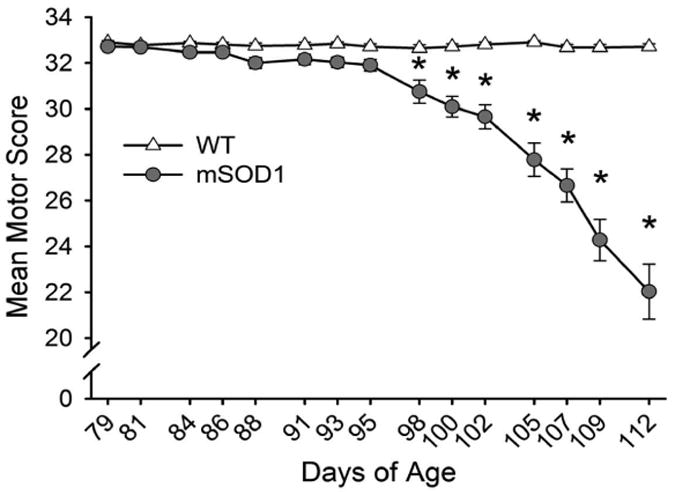
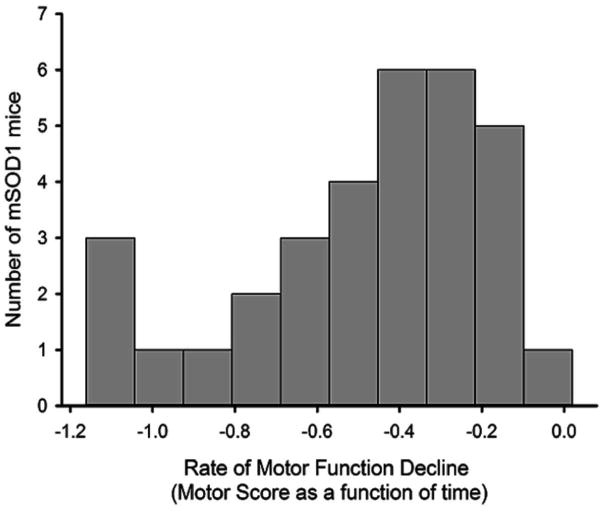
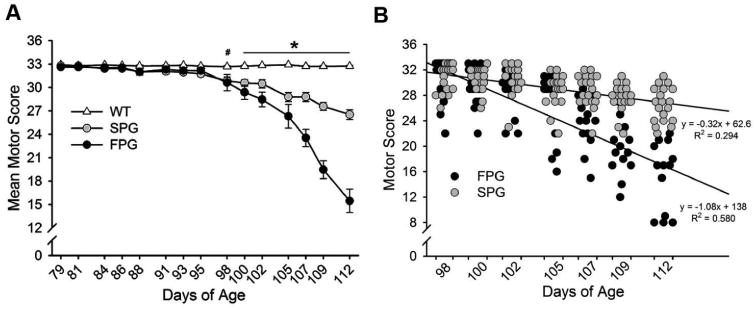
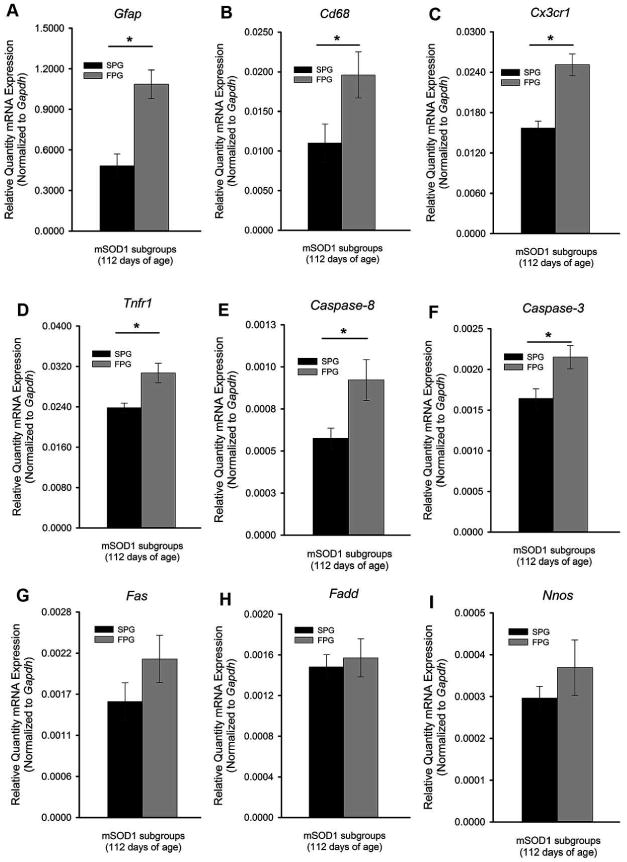
Disease progression rates among patients with amyotrophic lateral sclerosis (ALS) vary greatly. Although the majority of affected individuals survive 3-5 years following diagnosis, some subgroups experience a more rapidly progressing form, surviving less than 1 year, and other subgroups experience slowly progressing forms, surviving nearly 50 years. Genetic heterogeneity and environmental factors pose significant barriers in investigating patient progression rates. Similar to the case for humans, variation in survival within the mSOD1 mouse has been well documented, but different progression rates have not been investigated. The present study identifies two subgroups of B6SJL mSOD1(G93A) mice with different disease progression rates, a fast progression group (FPG) and slow progression group, as evidenced by differences in the rate of motor function decline. In addition, increased disease-associated gene expression within the FPG facial motor nucleus confirmed the presence of a more severe phenotype. We hypothesize that a more severe disease phenotype could be the result of 1) an earlier onset of axonal disconnection with a consistent degeneration rate or 2) a more severe or accelerated degenerative process. We performed a facial nerve transection axotomy in both mSOD1 subgroups prior to disease onset as a method to standardize the axonal disconnection. Instead of leading to comparable gene expression in both subgroups, this standardization did not eliminate the severe phenotype in the FPG facial nucleus, suggesting that the FPG phenotype is the result of a more severe or accelerated degenerative process. We theorize that these mSOD1 subgroups are representative of the rapid and slow disease phenotypes often experienced in ALS.
肌萎缩侧索硬化症(ALS)患者的疾病进展速度差异很大。虽然大多数患者在确诊后存活3至5年,但一些亚组经历进展更快的形式,存活时间不到1年,而其他亚组经历进展缓慢的形式,存活时间接近50年。基因异质性和环境因素在研究患者进展速度方面构成了重大障碍。与人类情况类似,mSOD1小鼠的存活差异已有充分记录,但不同的进展速度尚未得到研究。本研究确定了B6SJL mSOD1(G93A)小鼠的两个亚组,它们具有不同的疾病进展速度,即快速进展组(FPG)和缓慢进展组,运动功能下降速率的差异证明了这一点。此外,FPG面神经核内与疾病相关的基因表达增加证实了更严重表型的存在。我们假设更严重的疾病表型可能是以下两种情况的结果:1)轴突断开更早开始且退化速率一致,或者2)更严重或加速的退化过程。在疾病发作前,我们对两个mSOD1亚组都进行了面神经横断轴突切断术,作为使轴突断开标准化的一种方法。这种标准化并没有导致两个亚组的基因表达相当,反而没有消除FPG面神经核中的严重表型,这表明FPG表型是更严重或加速的退化过程的结果。我们推测这些mSOD1亚组代表了ALS中常见的快速和缓慢疾病表型。