Siretskiy Alexey, Sundqvist Tore, Voznesenskiy Mikhail, Spjuth Ola
Department of Information Technology, Uppsala University, P.O. Box 337, Uppsala, SE-75105 Sweden.
Department of Physical Chemistry, institute of Chemistry, St-Petersburg State University, Saint-Petersburg, Russia.
Gigascience. 2015 Jun 4;4:26. doi: 10.1186/s13742-015-0058-5. eCollection 2015.
New high-throughput technologies, such as massively parallel sequencing, have transformed the life sciences into a data-intensive field. The most common e-infrastructure for analyzing this data consists of batch systems that are based on high-performance computing resources; however, the bioinformatics software that is built on this platform does not scale well in the general case. Recently, the Hadoop platform has emerged as an interesting option to address the challenges of increasingly large datasets with distributed storage, distributed processing, built-in data locality, fault tolerance, and an appealing programming methodology.
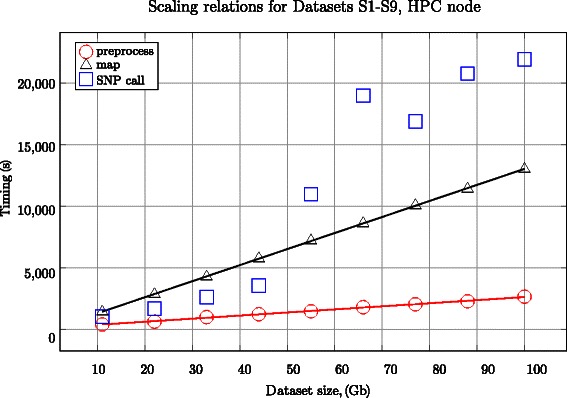
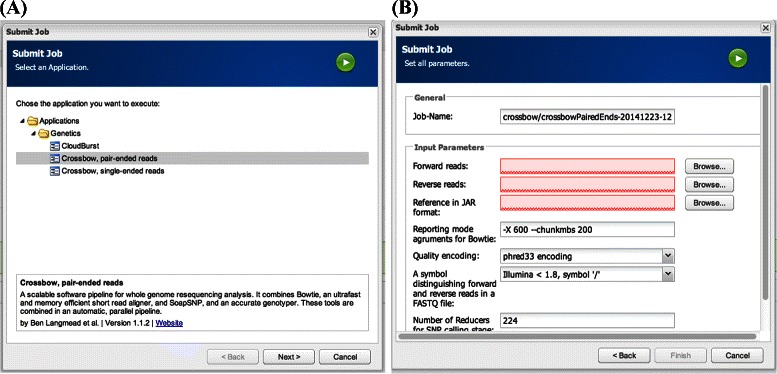
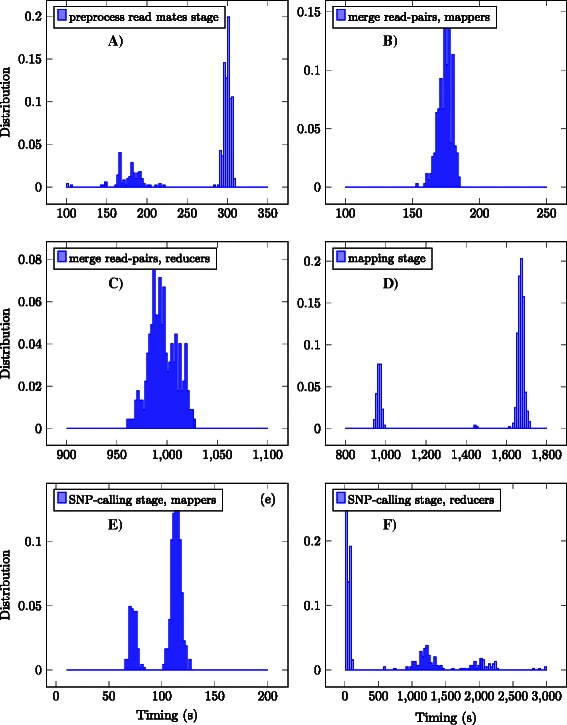
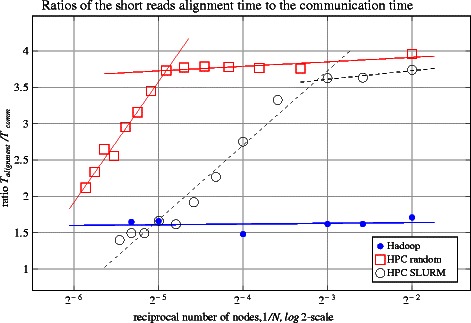
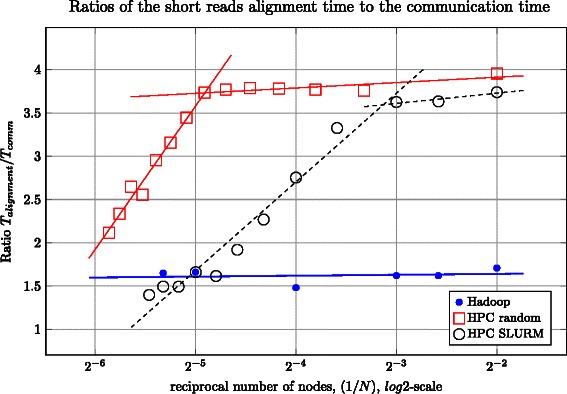
In this work we introduce metrics and report on a quantitative comparison between Hadoop and a single node of conventional high-performance computing resources for the tasks of short read mapping and variant calling. We calculate efficiency as a function of data size and observe that the Hadoop platform is more efficient for biologically relevant data sizes in terms of computing hours for both split and un-split data files. We also quantify the advantages of the data locality provided by Hadoop for NGS problems, and show that a classical architecture with network-attached storage will not scale when computing resources increase in numbers. Measurements were performed using ten datasets of different sizes, up to 100 gigabases, using the pipeline implemented in Crossbow. To make a fair comparison, we implemented an improved preprocessor for Hadoop with better performance for splittable data files. For improved usability, we implemented a graphical user interface for Crossbow in a private cloud environment using the CloudGene platform. All of the code and data in this study are freely available as open source in public repositories.
From our experiments we can conclude that the improved Hadoop pipeline scales better than the same pipeline on high-performance computing resources, we also conclude that Hadoop is an economically viable option for the common data sizes that are currently used in massively parallel sequencing. Given that datasets are expected to increase over time, Hadoop is a framework that we envision will have an increasingly important role in future biological data analysis.
新的高通量技术,如大规模平行测序,已将生命科学转变为一个数据密集型领域。用于分析此类数据的最常见电子基础设施由基于高性能计算资源的批处理系统组成;然而,构建在该平台上的生物信息学软件在一般情况下扩展性不佳。最近,Hadoop平台作为一种有趣的选择出现,可通过分布式存储、分布式处理、内置数据局部性、容错能力以及吸引人的编程方法来应对日益庞大的数据集所带来的挑战。
在本研究中,我们引入了指标,并报告了Hadoop与传统高性能计算资源的单个节点在短读段比对和变异检测任务上的定量比较。我们将效率计算为数据大小的函数,并观察到就分割和未分割数据文件的计算时长而言,Hadoop平台对于生物学相关数据大小更为高效。我们还量化了Hadoop为新一代测序问题提供的数据局部性优势,并表明当计算资源数量增加时,带有网络附属存储的经典架构将无法扩展。使用Crossbow中实现的流程,对多达100吉碱基的十个不同大小的数据集进行了测量。为了进行公平比较,我们为Hadoop实现了一个性能更好的改进型预处理器,用于可分割数据文件。为了提高可用性,我们在私有云环境中使用CloudGene平台为Crossbow实现了一个图形用户界面。本研究中的所有代码和数据均可在公共存储库中作为开源免费获取。
从我们的实验中可以得出结论,改进后的Hadoop流程比在高性能计算资源上运行的相同流程扩展性更好,我们还得出结论,对于当前大规模平行测序中使用的常见数据大小,Hadoop是一种经济可行的选择。鉴于数据集预计会随着时间增加,我们设想Hadoop框架在未来的生物数据分析中将发挥越来越重要的作用。