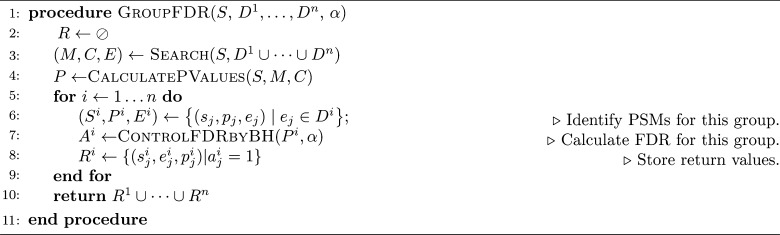Kertesz-Farkas Attila, Keich Uri, Noble William Stafford
Department of Genome Sciences, University of Washington, Seattle, Washington 98195, United States.
School of Mathematics and Statistics, University of Sydney, Camperdown, NSW 2006, Australia.
J Proteome Res. 2015 Aug 7;14(8):3027-38. doi: 10.1021/pr501173s. Epub 2015 Jun 30.
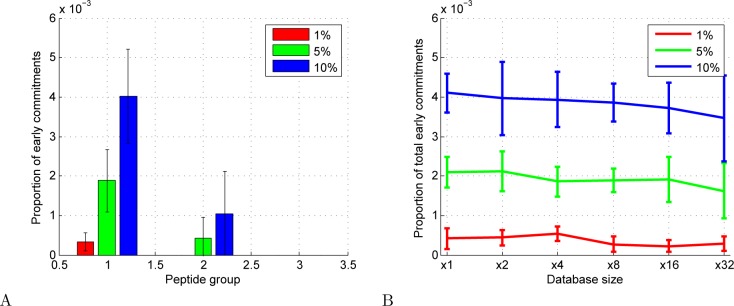
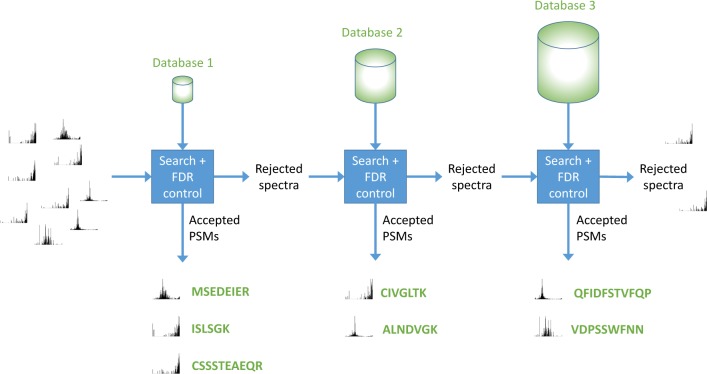
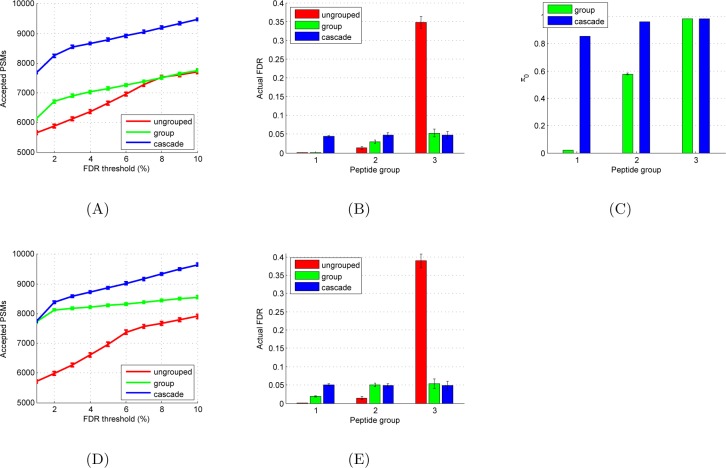
Accurate assignment of peptide sequences to observed fragmentation spectra is hindered by the large number of hypotheses that must be considered for each observed spectrum. A high score assigned to a particular peptide-spectrum match (PSM) may not end up being statistically significant after multiple testing correction. Researchers can mitigate this problem by controlling the hypothesis space in various ways: considering only peptides resulting from enzymatic cleavages, ignoring possible post-translational modifications or single nucleotide variants, etc. However, these strategies sacrifice identifications of spectra generated by rarer types of peptides. In this work, we introduce a statistical testing framework, cascade search, that directly addresses this problem. The method requires that the user specify a priori a statistical confidence threshold as well as a series of peptide databases. For instance, such a cascade of databases could include fully tryptic, semitryptic, and nonenzymatic peptides or peptides with increasing numbers of modifications. Cascaded search then gradually expands the list of candidate peptides from more likely peptides toward rare peptides, sequestering at each stage any spectrum that is identified with a specified statistical confidence. We compare cascade search to a standard procedure that lumps all of the peptides into a single database, as well as to a previously described group FDR procedure that computes the FDR separately within each database. We demonstrate, using simulated and real data, that cascade search identifies more spectra at a fixed FDR threshold than with either the ungrouped or grouped approach. Cascade search thus provides a general method for maximizing the number of identified spectra in a statistically rigorous fashion.
将肽序列准确地分配到观察到的碎片光谱中,会受到大量假设的阻碍,因为对于每个观察到的光谱都必须考虑这些假设。在进行多重检验校正后,赋予特定肽段 - 光谱匹配(PSM)的高分最终可能不具有统计学意义。研究人员可以通过多种方式控制假设空间来缓解这个问题:仅考虑酶切产生的肽段,忽略可能的翻译后修饰或单核苷酸变体等。然而,这些策略牺牲了对由稀有类型肽段产生的光谱的鉴定。在这项工作中,我们引入了一种统计检验框架——级联搜索,它直接解决了这个问题。该方法要求用户事先指定一个统计置信阈值以及一系列肽数据库。例如,这样的数据库级联可以包括完全胰蛋白酶酶切的、半胰蛋白酶酶切的和非酶切的肽段,或者修饰数量不断增加的肽段。然后,级联搜索从更可能的肽段逐渐扩展候选肽段列表,直至稀有肽段,并在每个阶段隔离任何以指定统计置信度鉴定出的光谱。我们将级联搜索与一种将所有肽段集中到单个数据库中的标准程序进行比较,同时也与之前描述的在每个数据库中单独计算错误发现率(FDR)的分组FDR程序进行比较。我们使用模拟数据和真实数据证明,在固定的FDR阈值下,级联搜索比未分组或分组方法鉴定出的光谱更多。因此,级联搜索提供了一种以统计严谨的方式最大化鉴定光谱数量的通用方法。