Carbonara Roberta, Carocci Alessia, Roussel Julien, Crescenzo Giuseppe, Buonavoglia Canio, Franchini Carlo, Lentini Giovanni, Camerino Diana Conte, Desaphy Jean-François
Section of Pharmacology, Department of Pharmacy & Drug Sciences, University of Bari Aldo Moro Bari, Italy.
Section of Medicinal Chemistry, Department of Pharmacy & Drug Sciences, University of Bari Aldo Moro Bari, Italy.
Front Pharmacol. 2015 Jul 24;6:155. doi: 10.3389/fphar.2015.00155. eCollection 2015.
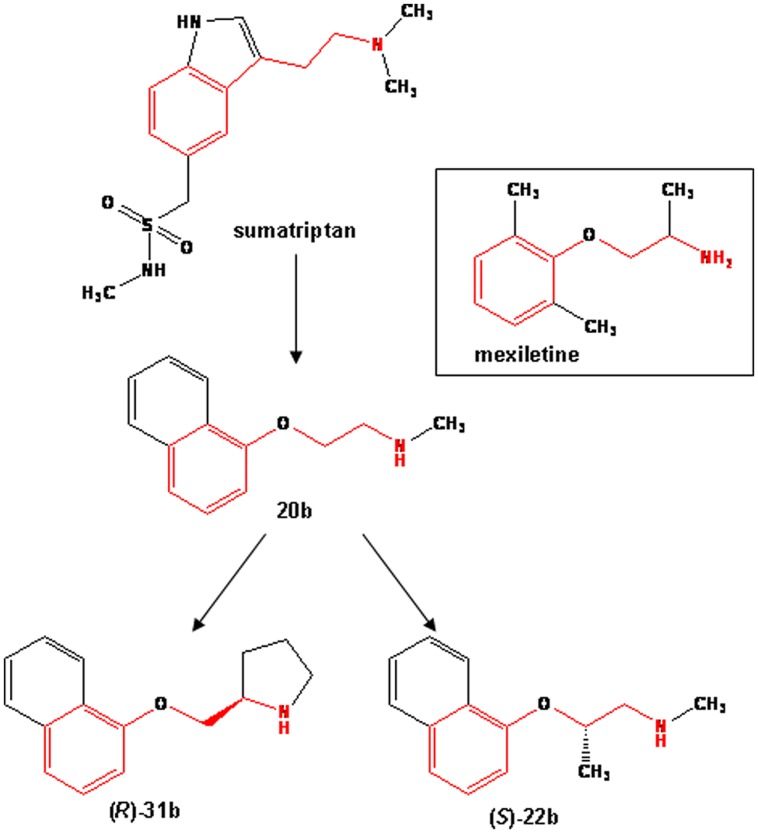
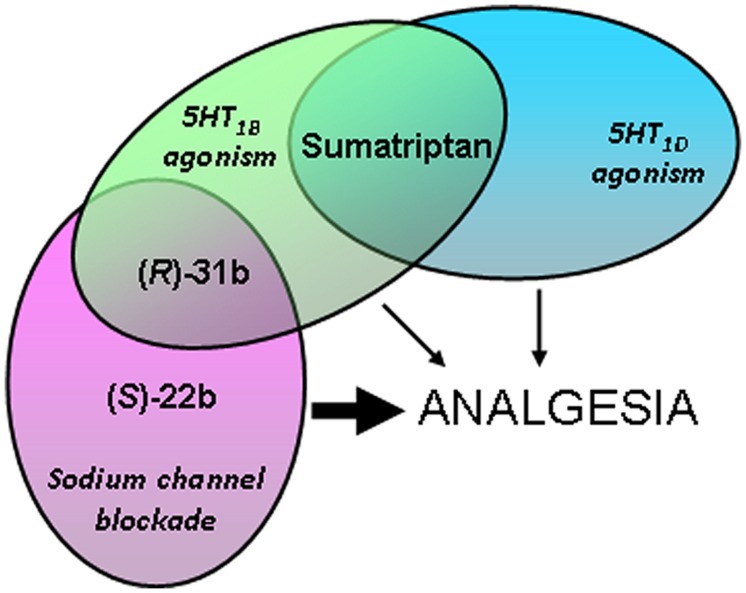
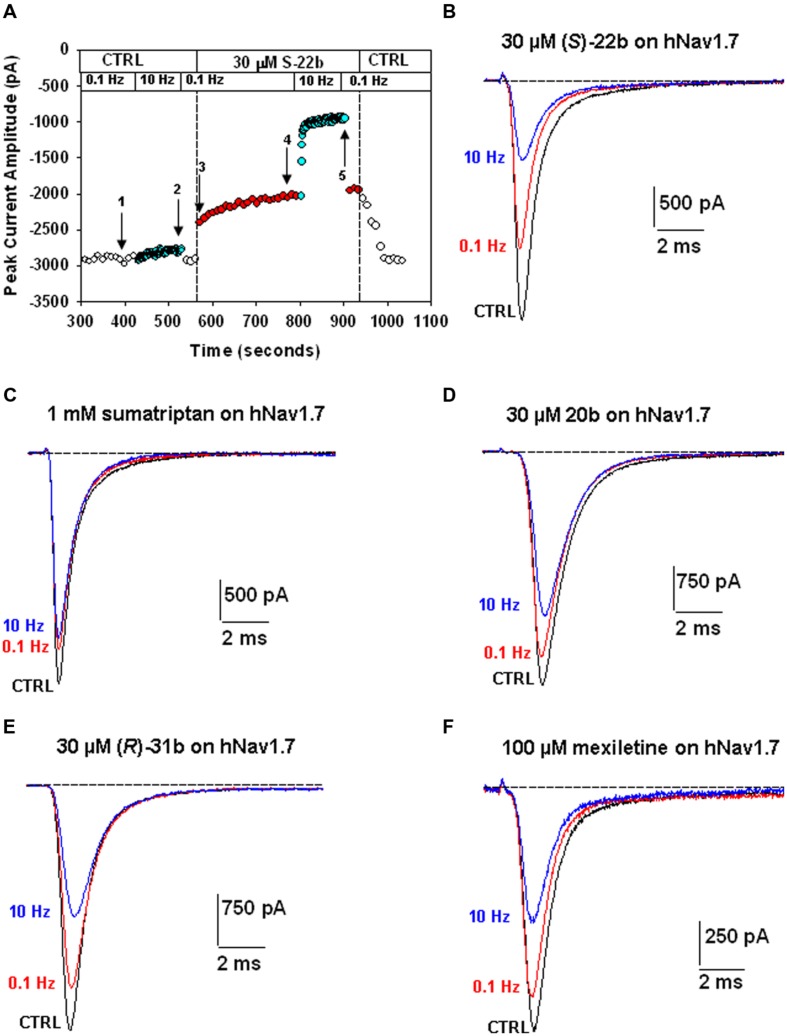
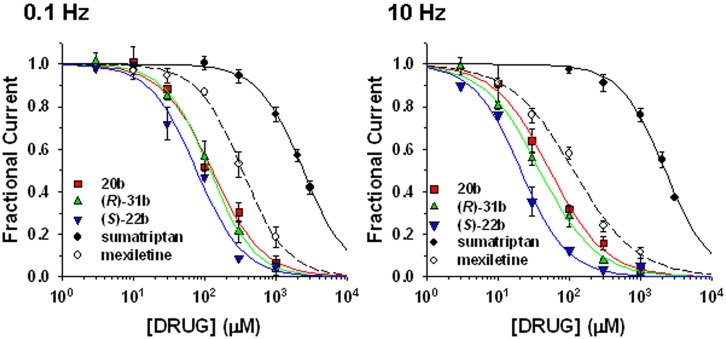
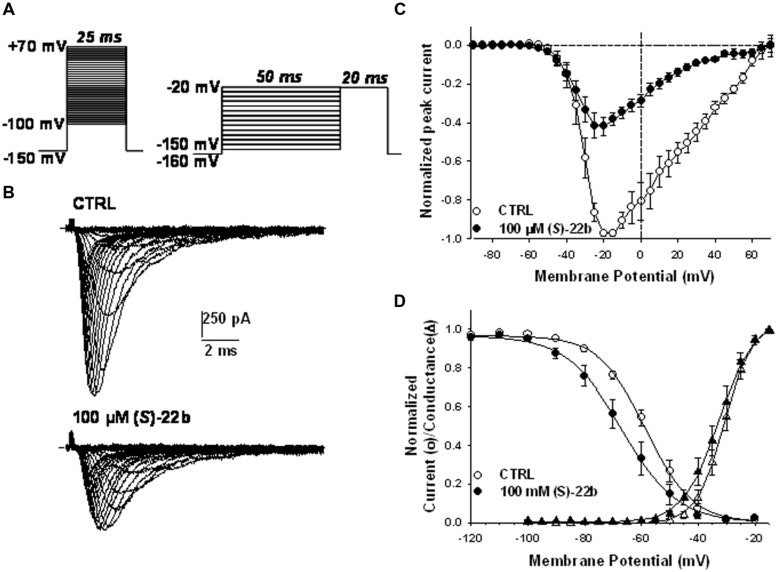
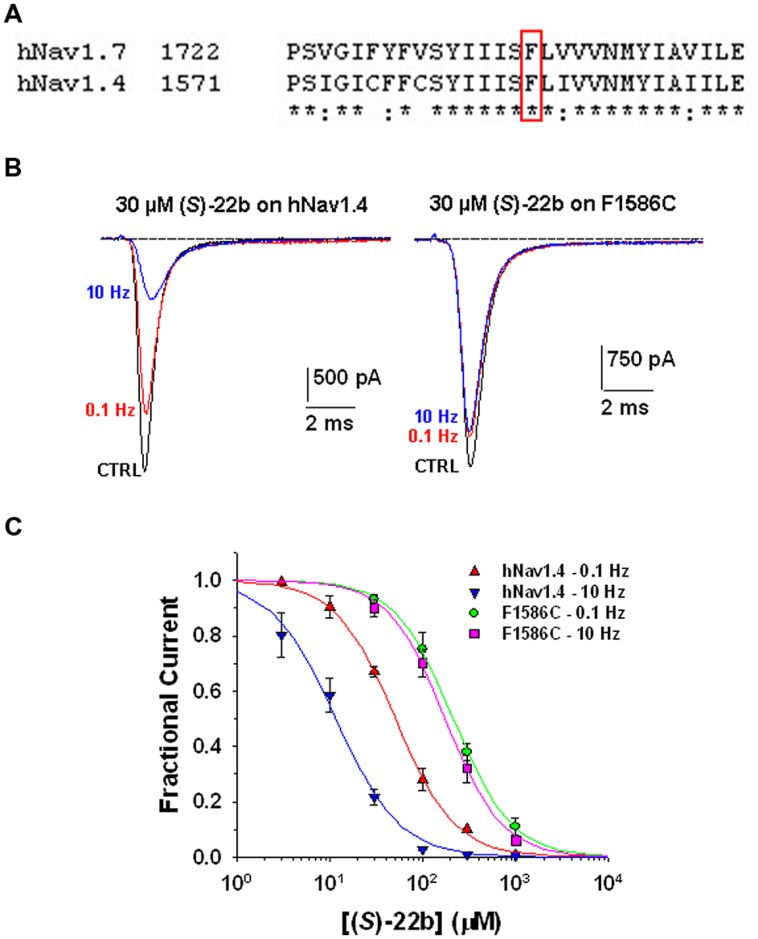
Voltage-gated sodium channels are known to play a pivotal role in perception and transmission of pain sensations. Gain-of-function mutations in the genes encoding the peripheral neuronal sodium channels, hNav1.7-1.9, cause human painful diseases. Thus while treatment of chronic pain remains an unmet clinical need, sodium channel blockers are considered as promising druggable targets. In a previous study, we evaluated the analgesic activity of sumatriptan, an agonist of serotonin 5HT1B/D receptors, and some new chiral bioisosteres, using the hot plate test in the mouse. Interestingly, we observed that the analgesic effectiveness was not necessarily correlated to serotonin agonism. In this study, we evaluated whether sumatriptan and its congeners may inhibit heterologously expressed hNav1.7 sodium channels using the patch-clamp method. We show that sumatriptan blocks hNav1.7 channels only at very high, supratherapeutic concentrations. In contrast, its three analogs, namely 20b, (R)-31b, and (S)-22b, exert a dose and use-dependent sodium channel block. At 0.1 and 10 Hz stimulation frequencies, the most potent compound, (S)-22b, was 4.4 and 1.7 fold more potent than the well-known sodium channel blocker mexiletine. The compound induces a negative shift of voltage dependence of fast inactivation, suggesting higher affinity to the inactivated channel. Accordingly, we show that (S)-22b likely binds the conserved local anesthetic receptor within voltage-gated sodium channels. Combining these results with the previous ones, we hypothesize that use-dependent sodium channel blockade contributes to the analgesic activity of (R)-31b and (S)-22b. These later compounds represent promising lead compounds for the development of efficient analgesics, the mechanism of action of which may include a dual action on sodium channels and 5HT1D receptors.
已知电压门控钠通道在痛觉的感知和传递中起关键作用。编码外周神经元钠通道hNav1.7 - 1.9的基因功能获得性突变会导致人类疼痛性疾病。因此,尽管慢性疼痛的治疗仍是未满足的临床需求,但钠通道阻滞剂被认为是有前景的可成药靶点。在之前的一项研究中,我们使用小鼠热板试验评估了5-羟色胺1B/D受体激动剂舒马曲坦及一些新的手性生物电子等排体的镇痛活性。有趣的是,我们观察到镇痛效果不一定与5-羟色胺激动作用相关。在本研究中,我们使用膜片钳方法评估了舒马曲坦及其同系物是否能抑制异源表达的hNav1.7钠通道。我们发现舒马曲坦仅在非常高的、超治疗浓度下才会阻断hNav1.7通道。相比之下,它的三个类似物,即20b、(R)-31b和(S)-22b,呈现出剂量和使用依赖性的钠通道阻断作用。在0.1和10 Hz刺激频率下,最有效的化合物(S)-22b比著名的钠通道阻滞剂美西律分别强4.4倍和1.7倍。该化合物使快速失活的电压依赖性产生负向偏移,表明其对失活通道具有更高的亲和力。因此,我们表明(S)-22b可能与电压门控钠通道内保守的局部麻醉药受体结合。将这些结果与之前的结果相结合,我们推测使用依赖性钠通道阻断作用有助于(R)-31b和(S)-22b的镇痛活性。这些后期化合物是开发高效镇痛药的有前景的先导化合物,其作用机制可能包括对钠通道和5HT1D受体的双重作用。