Kumar Praveen Kumar Raj, Hoang Thanh V, Robinson Michael L, Tsonis Panagiotis A, Liang Chun
Department of Biology, Miami University, Oxford, Ohio 45056, USA.
Department of Biology and Center for Tissue Regeneration and Engineering, The University of Dayton, Dayton, OH 45469.
Sci Rep. 2015 Aug 25;5:13443. doi: 10.1038/srep13443.
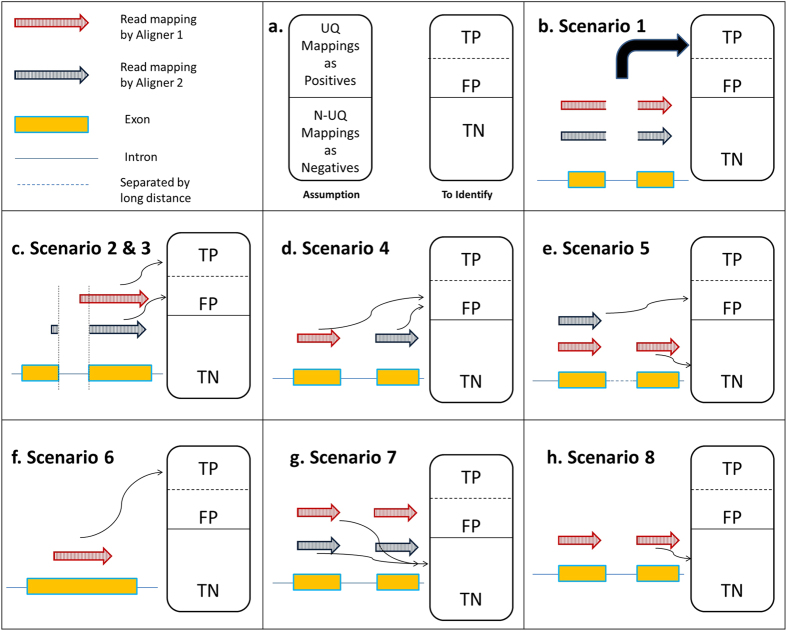
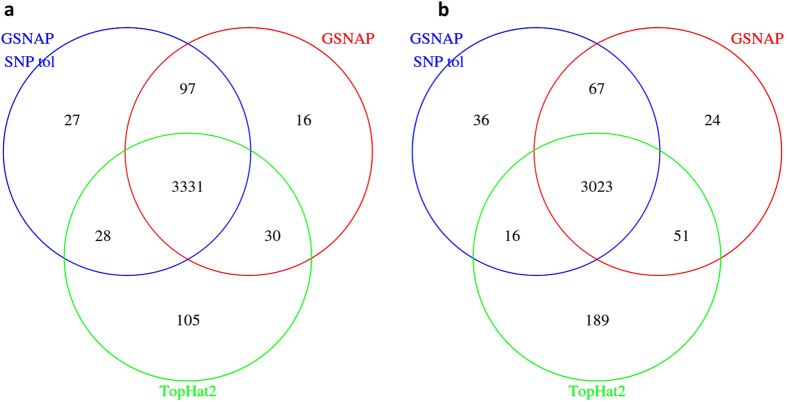
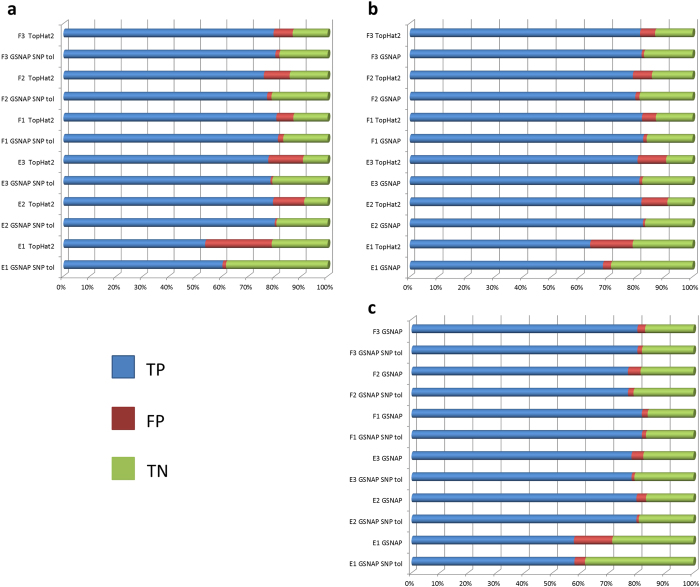
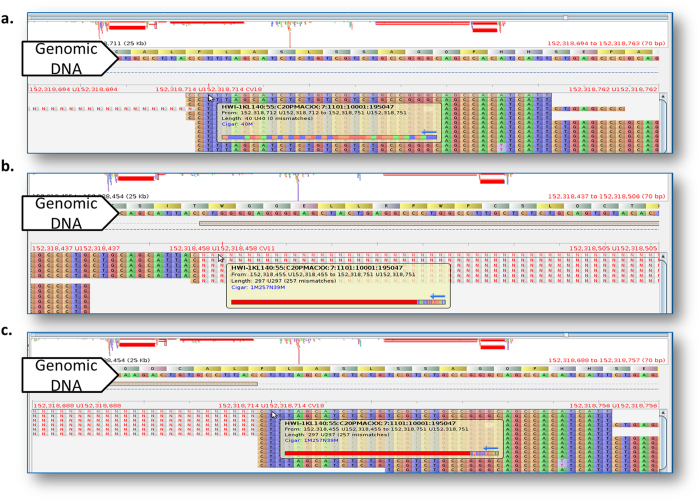
The fundamental task in RNA-Seq-based transcriptome analysis is alignment of millions of short reads to the reference genome or transcriptome. Choosing the right tool for the dataset in hand from many existent RNA-Seq alignment packages remains a critical challenge for downstream analysis. To facilitate this choice, we designed a novel tool for comparing alignment results of user data based on the relative reliability of uniquely aligned reads (CADBURE). CADBURE can easily evaluate different aligners, or different parameter sets using the same aligner, and selects the best alignment result for any RNA-Seq dataset. Strengths of CADBURE include the ability to compare alignment results without the need for synthetic data such as simulated genomes, alignment regeneration and randomly subsampled datasets. The benefit of a CADBURE selected alignment result was supported by differentially expressed gene (DEG) analysis. We demonstrated that the use of CADBURE to select the best alignment from a number of different alignment results could change the number of DEGs by as much as 10%. In particular, the CADBURE selected alignment result favors fewer false positives in the DEG analysis. We also verified differential expression of eighteen genes with RT-qPCR validation experiments. CADBURE is an open source tool (http://cadbure.sourceforge.net/).
基于RNA测序的转录组分析的基本任务是将数百万条短读段比对到参考基因组或转录组上。从众多现有的RNA测序比对软件包中为手头的数据集选择合适的工具,对于下游分析而言仍然是一项严峻的挑战。为便于做出这种选择,我们基于唯一比对读段的相对可靠性设计了一种用于比较用户数据比对结果的新型工具(CADBURE)。CADBURE能够轻松评估不同的比对器,或者使用同一比对器的不同参数集,并为任何RNA测序数据集选择最佳比对结果。CADBURE的优势包括无需诸如模拟基因组、比对重新生成和随机抽样数据集等合成数据即可比较比对结果。差异表达基因(DEG)分析支持了CADBURE选择的比对结果的优势。我们证明,使用CADBURE从多个不同比对结果中选择最佳比对,可使差异表达基因的数量变化多达10%。特别是,CADBURE选择的比对结果在DEG分析中有利于减少假阳性。我们还通过逆转录定量PCR验证实验证实了18个基因的差异表达。CADBURE是一个开源工具(http://cadbure.sourceforge.net/)。