Elbl Paula, Navarro Bruno V, de Oliveira Leandro F, Almeida Juliana, Mosini Amanda C, Dos Santos André L W, Rossi Magdalena, Floh Eny I S
Departamento de Botânica, Instituto de Biociências, Universidade de São Paulo, São Paulo, Brasil.
PLoS One. 2015 Aug 27;10(8):e0136714. doi: 10.1371/journal.pone.0136714. eCollection 2015.
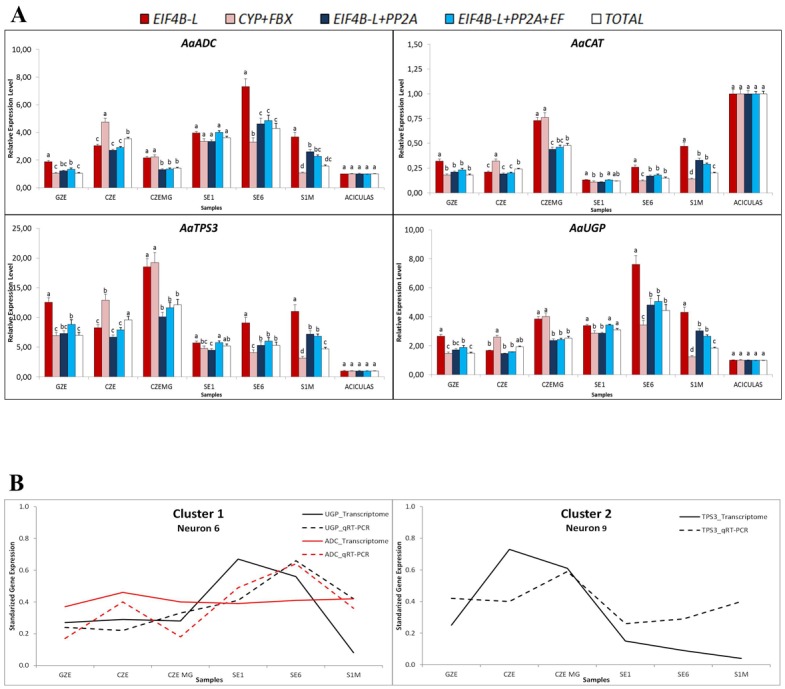
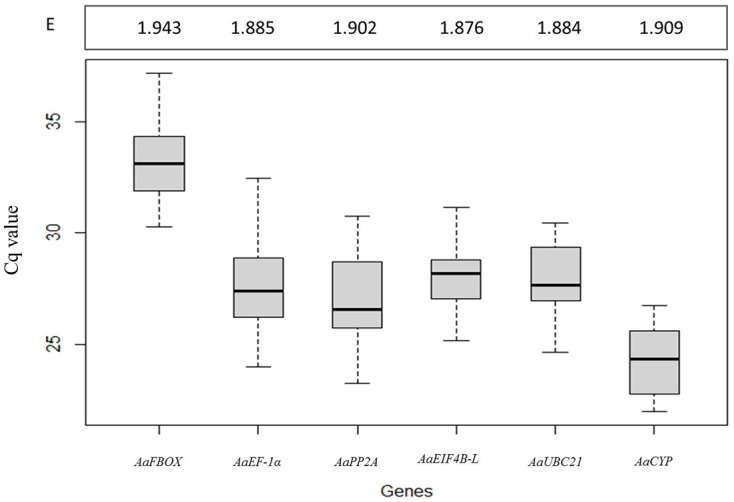
Quantitative analysis of gene expression is a fundamental experimental approach in many fields of plant biology, but it requires the use of internal controls representing constitutively expressed genes for reliable transcript quantification. In this study, we identified fifteen putative reference genes from an A. angustifolia transcriptome database. Variation in transcript levels was first evaluated in silico by comparing read counts and then by quantitative real-time PCR (qRT-PCR), resulting in the identification of six candidate genes. The consistency of transcript abundance was also calculated applying geNorm and NormFinder software packages followed by a validation approach using four target genes. The results presented here indicate that a diverse set of samples should ideally be used in order to identify constitutively expressed genes, and that the use of any two reference genes in combination, of the six tested genes, is sufficient for effective expression normalization. Finally, in agreement with the in silico prediction, a comprehensive analysis of the qRT-PCR data combined with validation analysis revealed that AaEIF4B-L and AaPP2A are the most suitable reference genes for comparative studies of A. angustifolia gene expression.
基因表达的定量分析是植物生物学许多领域的一种基本实验方法,但它需要使用代表组成型表达基因的内参来进行可靠的转录本定量。在本研究中,我们从狭叶南洋杉转录组数据库中鉴定出15个假定的参考基因。首先通过比较读数计数在计算机上评估转录本水平的变化,然后通过定量实时PCR(qRT-PCR)进行评估,从而鉴定出6个候选基因。还应用geNorm和NormFinder软件包计算转录本丰度的一致性,随后使用4个靶基因进行验证。这里给出的结果表明,理想情况下应该使用多样的样本集来鉴定组成型表达基因,并且在6个测试基因中,任意两个参考基因组合使用就足以进行有效的表达归一化。最后,与计算机预测一致,对qRT-PCR数据的综合分析与验证分析表明,AaEIF4B-L和AaPP2A是狭叶南洋杉基因表达比较研究中最合适的参考基因。