Yigit Gökhan, Brown Karen E, Kayserili Hülya, Pohl Esther, Caliebe Almuth, Zahnleiter Diana, Rosser Elisabeth, Bögershausen Nina, Uyguner Zehra Oya, Altunoglu Umut, Nürnberg Gudrun, Nürnberg Peter, Rauch Anita, Li Yun, Thiel Christian Thomas, Wollnik Bernd
Institute of Human Genetics, University of Cologne Cologne, Germany ; Center for Molecular Medicine Cologne (CMMC), University of Cologne Cologne, Germany ; Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), University of Cologne Cologne, Germany.
Chromosome Biology Group, MRC Clinical Sciences Centre, Imperial College School of Medicine, Hammersmith Hospital London, W12 0NN, UK.
Mol Genet Genomic Med. 2015 Sep;3(5):467-80. doi: 10.1002/mgg3.158. Epub 2015 May 24.
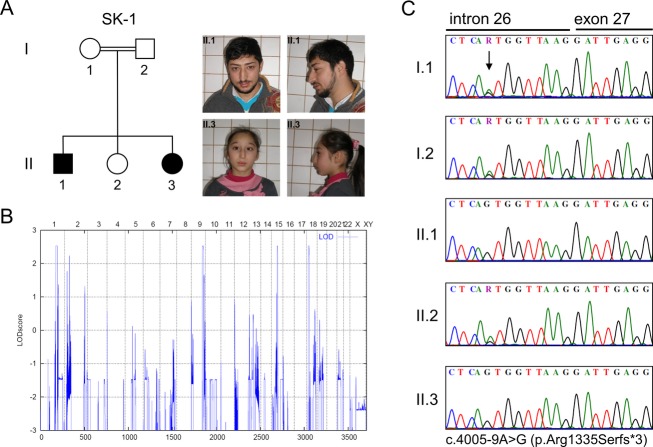
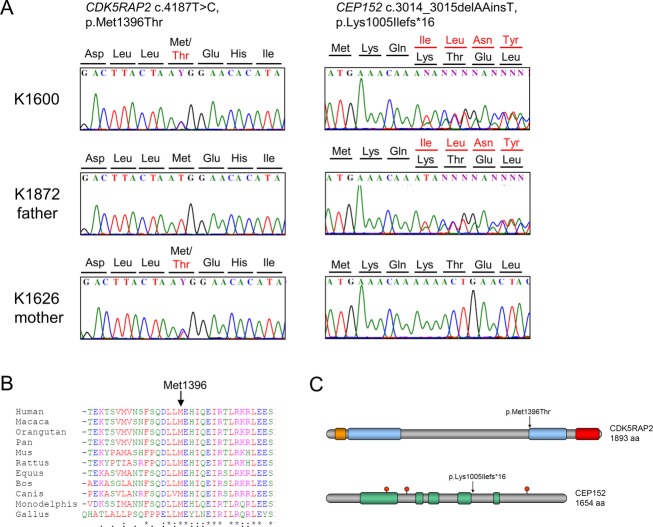
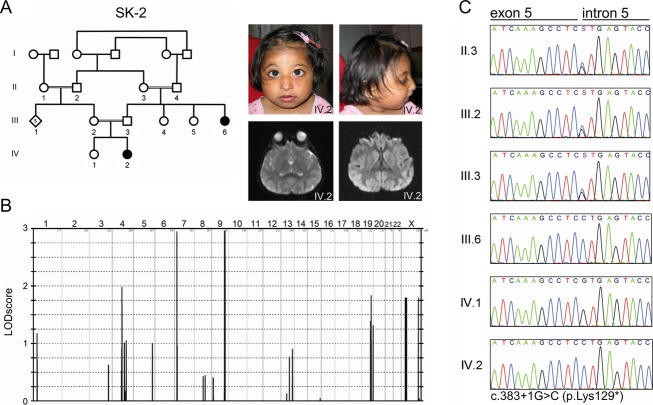
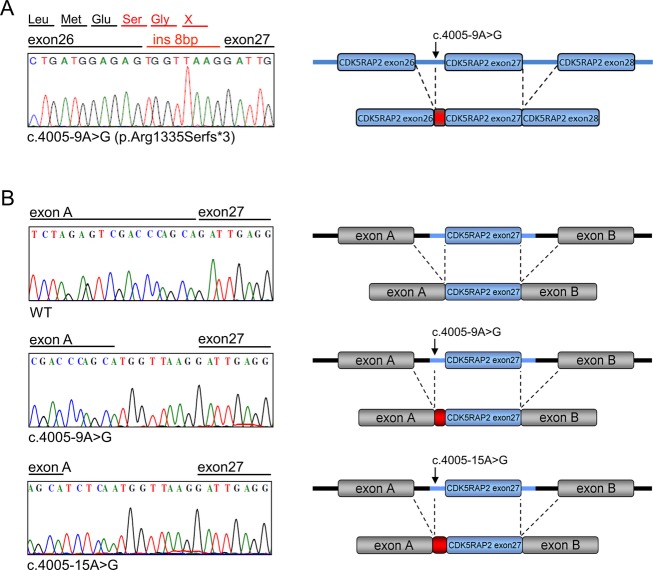
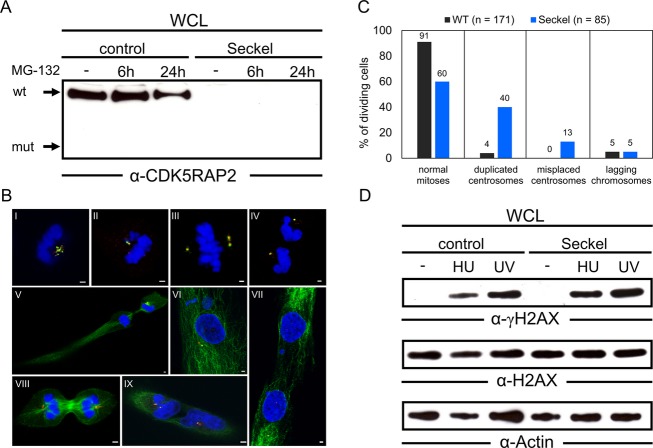
Seckel syndrome is a heterogeneous, autosomal recessive disorder marked by prenatal proportionate short stature, severe microcephaly, intellectual disability, and characteristic facial features. Here, we describe the novel homozygous splice-site mutations c.383+1G>C and c.4005-9A>G in CDK5RAP2 in two consanguineous families with Seckel syndrome. CDK5RAP2 (CEP215) encodes a centrosomal protein which is known to be essential for centrosomal cohesion and proper spindle formation and has been shown to be causally involved in autosomal recessive primary microcephaly. We establish CDK5RAP2 as a disease-causing gene for Seckel syndrome and show that loss of functional CDK5RAP2 leads to severe defects in mitosis and spindle organization, resulting in cells with abnormal nuclei and centrosomal pattern, which underlines the important role of centrosomal and mitotic proteins in the pathogenesis of the disease. Additionally, we present an intriguing case of possible digenic inheritance in Seckel syndrome: A severely affected child of nonconsanguineous German parents was found to carry heterozygous mutations in CDK5RAP2 and CEP152. This finding points toward a potential additive genetic effect of mutations in CDK5RAP2 and CEP152.
塞克尔综合征是一种异质性常染色体隐性疾病,其特征为出生前身材成比例矮小、严重小头畸形、智力障碍以及特殊面容。在此,我们描述了两个患塞克尔综合征的近亲家庭中,CDK5RAP2基因存在新的纯合剪接位点突变c.383+1G>C和c.4005-9A>G。CDK5RAP2(CEP215)编码一种中心体蛋白,已知该蛋白对于中心体黏附及纺锤体正常形成至关重要,并且已证明其与常染色体隐性原发性小头畸形存在因果关系。我们确定CDK5RAP2是塞克尔综合征的致病基因,并表明功能性CDK5RAP2的缺失会导致有丝分裂和纺锤体组织出现严重缺陷,从而产生细胞核和中心体模式异常的细胞,这突出了中心体和有丝分裂蛋白在该疾病发病机制中的重要作用。此外,我们还展示了一个塞克尔综合征可能存在双基因遗传的有趣病例:一对非近亲的德国父母所生的一名严重患病儿童,被发现其CDK5RAP2和CEP152基因携带杂合突变。这一发现表明CDK5RAP2和CEP152基因的突变可能存在潜在的累加遗传效应。