Adato Orit, Ninyo Noga, Gophna Uri, Snir Sagi
Department of Evolutionary Biology, University of Haifa, Haifa, Israel.
Department of Molecular Microbiology and Biotechnology Tel Aviv University, Tel-Aviv, Israel.
PLoS Comput Biol. 2015 Oct 6;11(10):e1004408. doi: 10.1371/journal.pcbi.1004408. eCollection 2015 Oct.
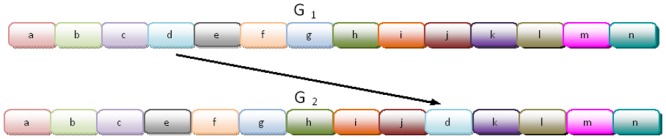
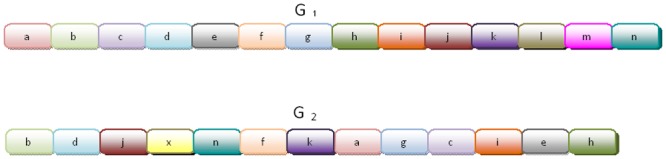
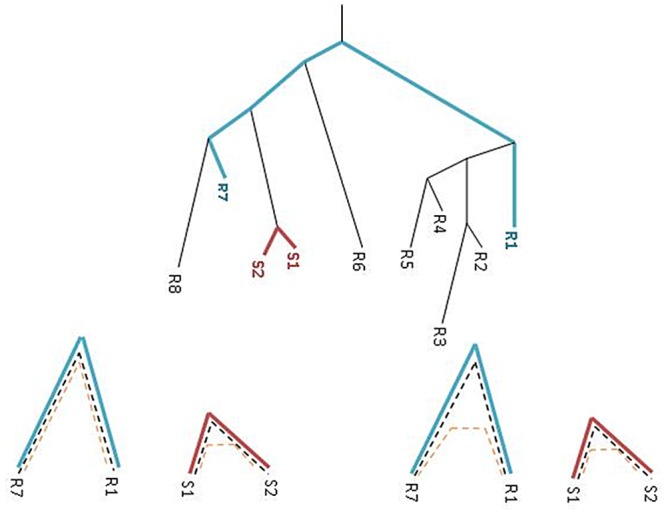
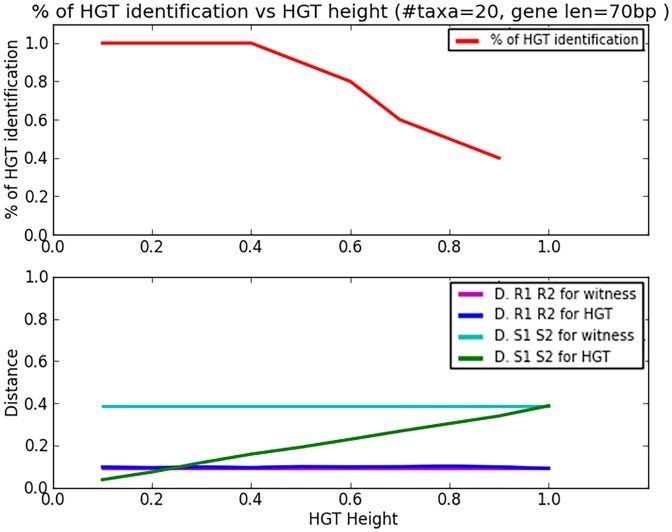
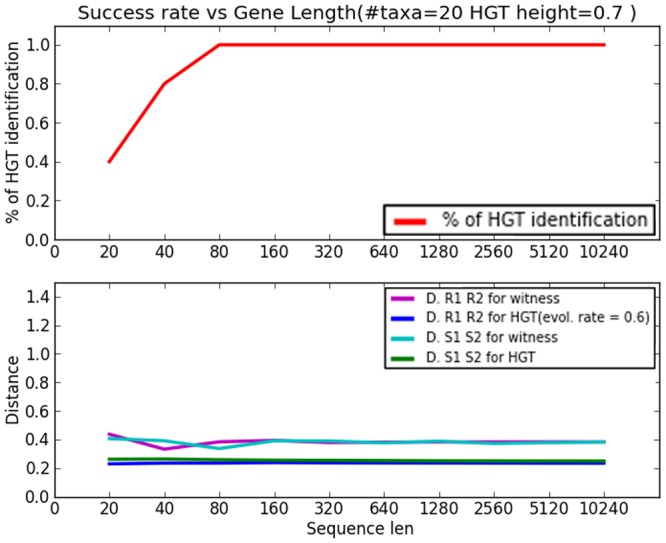
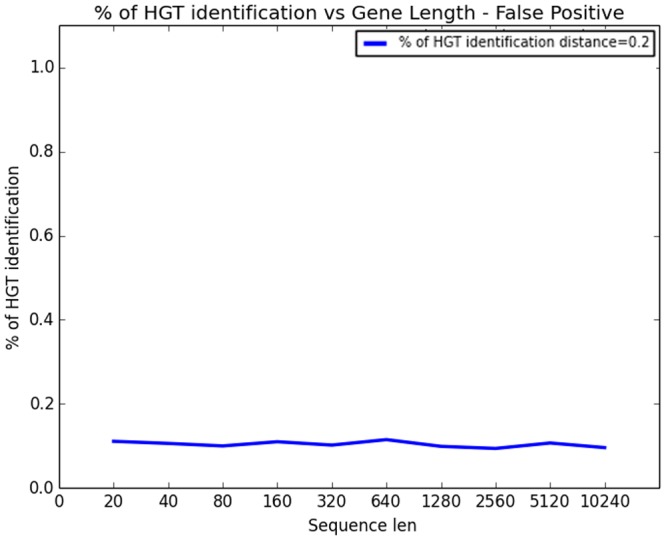
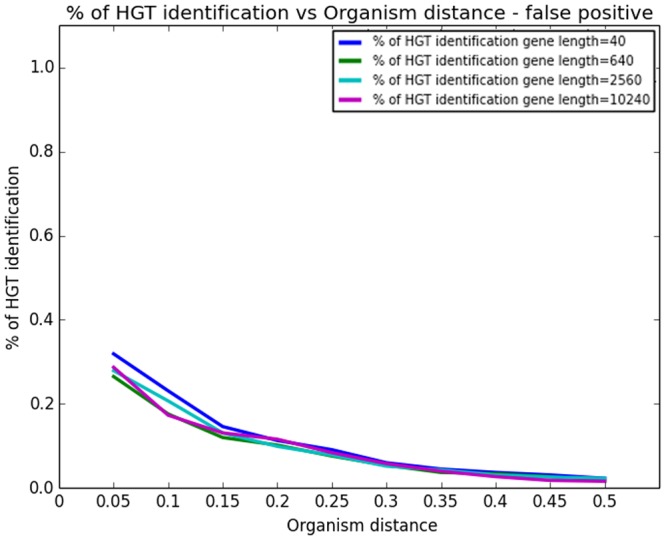
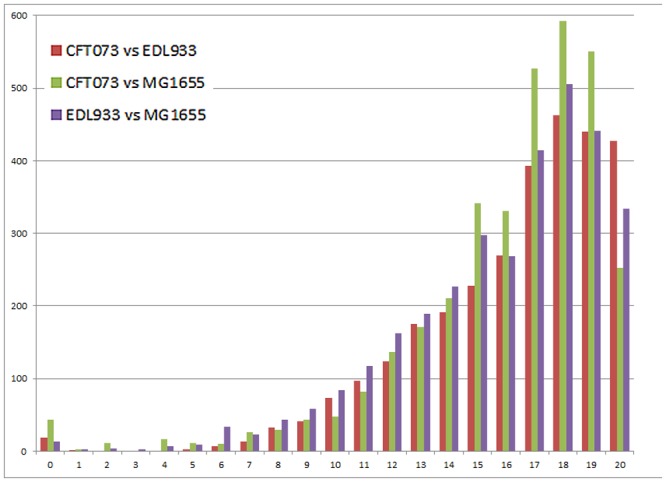
Horizontal gene transfer (HGT), the transfer of genetic material between organisms, is crucial for genetic innovation and the evolution of genome architecture. Existing HGT detection algorithms rely on a strong phylogenetic signal distinguishing the transferred sequence from ancestral (vertically derived) genes in its recipient genome. Detecting HGT between closely related species or strains is challenging, as the phylogenetic signal is usually weak and the nucleotide composition is normally nearly identical. Nevertheless, there is a great importance in detecting HGT between congeneric species or strains, especially in clinical microbiology, where understanding the emergence of new virulent and drug-resistant strains is crucial, and often time-sensitive. We developed a novel, self-contained technique named Near HGT, based on the synteny index, to measure the divergence of a gene from its native genomic environment and used it to identify candidate HGT events between closely related strains. The method confirms candidate transferred genes based on the constant relative mutability (CRM). Using CRM, the algorithm assigns a confidence score based on "unusual" sequence divergence. A gene exhibiting exceptional deviations according to both synteny and mutability criteria, is considered a validated HGT product. We first employed the technique to a set of three E. coli strains and detected several highly probable horizontally acquired genes. We then compared the method to existing HGT detection tools using a larger strain data set. When combined with additional approaches our new algorithm provides richer picture and brings us closer to the goal of detecting all newly acquired genes in a particular strain.
水平基因转移(HGT),即生物体之间遗传物质的转移,对于遗传创新和基因组结构的进化至关重要。现有的HGT检测算法依赖于强大的系统发育信号,以将转移的序列与其受体基因组中的祖先(垂直衍生)基因区分开来。在密切相关的物种或菌株之间检测HGT具有挑战性,因为系统发育信号通常较弱,且核苷酸组成通常几乎相同。然而,在同属物种或菌株之间检测HGT非常重要,尤其是在临床微生物学中,了解新的致病和耐药菌株的出现至关重要,而且往往对时间敏感。我们基于共线性指数开发了一种名为Near HGT的新颖、独立的技术,以测量基因与其原生基因组环境的差异,并使用它来识别密切相关菌株之间的候选HGT事件。该方法基于恒定相对突变率(CRM)来确认候选转移基因。利用CRM,该算法根据“异常”的序列差异分配一个置信度得分。根据共线性和突变标准表现出异常偏差的基因被视为经过验证的HGT产物。我们首先将该技术应用于一组三株大肠杆菌,并检测到几个极有可能通过水平方式获得的基因。然后,我们使用更大的菌株数据集将该方法与现有的HGT检测工具进行比较。当与其他方法结合使用时,我们的新算法能提供更丰富的信息,并使我们更接近检测特定菌株中所有新获得基因的目标。