Lange Anja, Jost Steffen, Heider Dominik, Bock Christina, Budeus Bettina, Schilling Elmar, Strittmatter Axel, Boenigk Jens, Hoffmann Daniel
Research Group Bioinformatics, Faculty of Biology, University of Duisburg-Essen, Essen, Germany.
Department of Biodiversity, Faculty of Biology, University of Duisburg-Essen, Essen, Germany.
PLoS One. 2015 Nov 2;10(11):e0141590. doi: 10.1371/journal.pone.0141590. eCollection 2015.
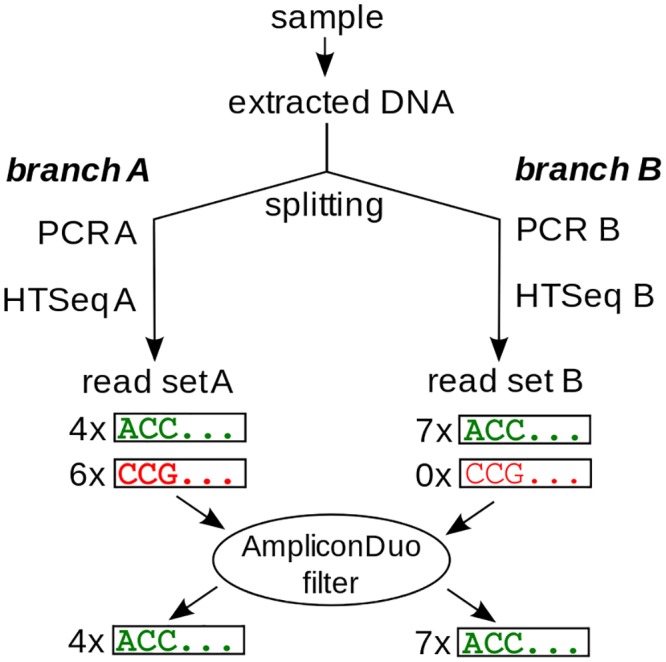
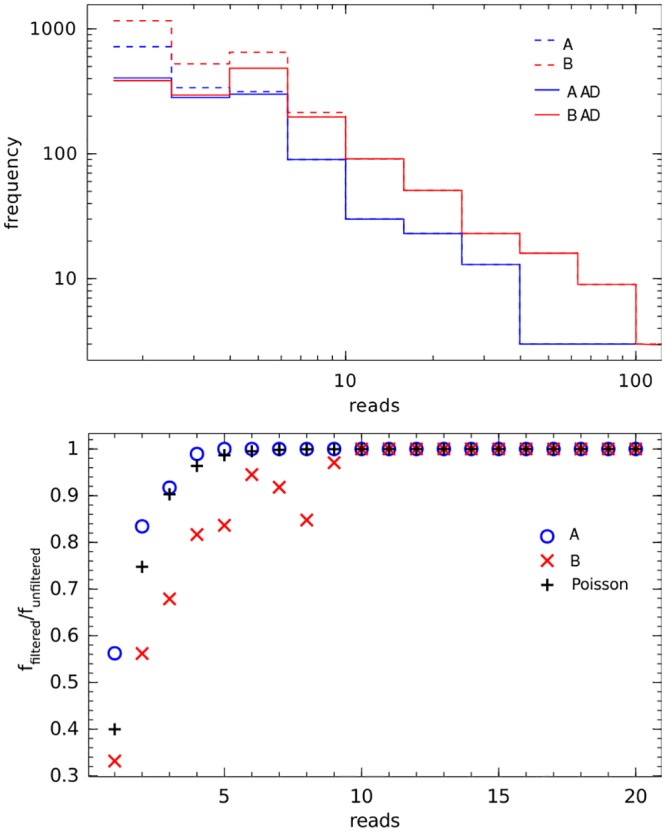
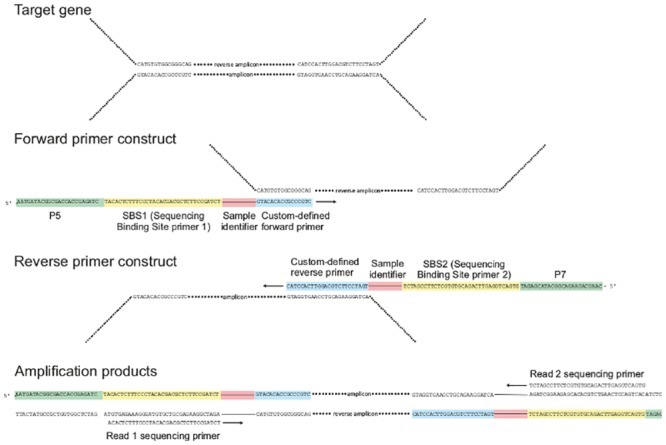
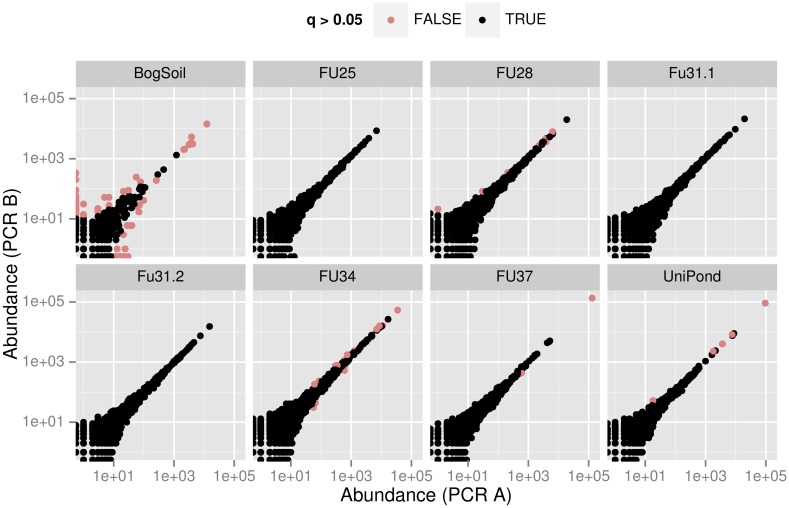
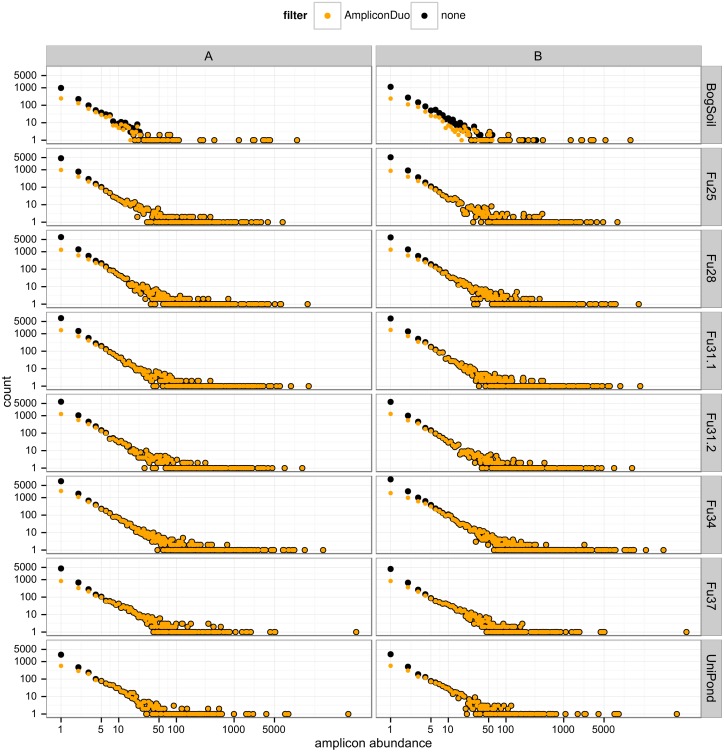
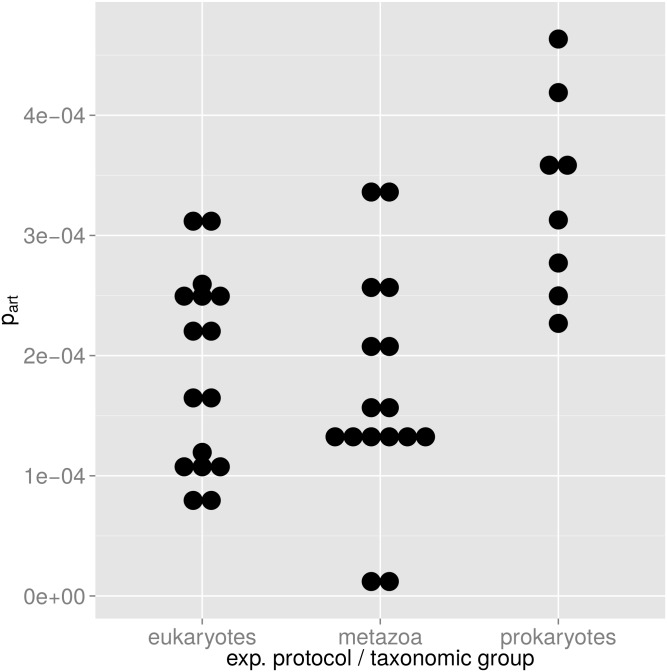
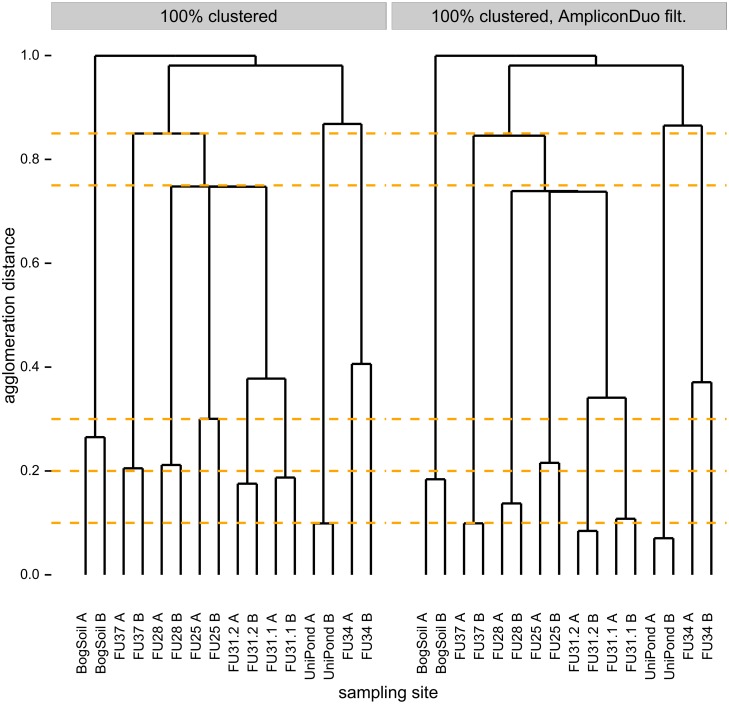
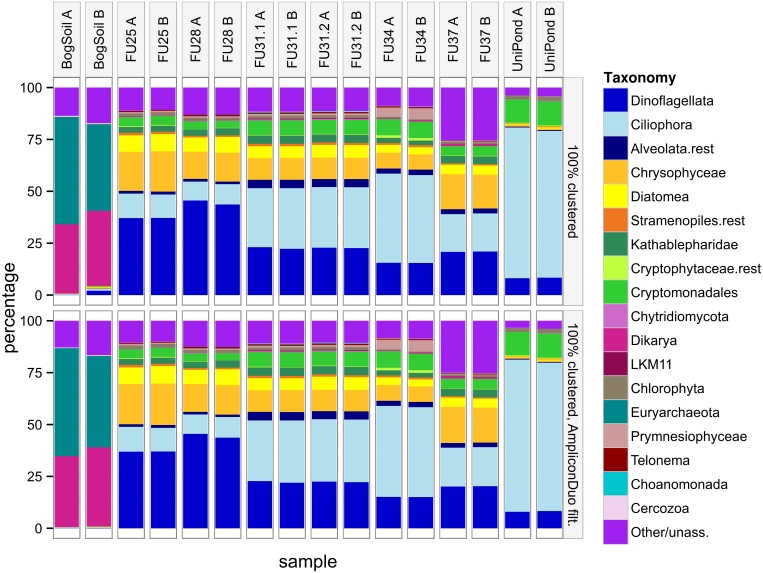
High throughput sequencing (HTSeq) of small ribosomal subunit amplicons has the potential for a comprehensive characterization of microbial community compositions, down to rare species. However, the error-prone nature of the multi-step experimental process requires that the resulting raw sequences are subjected to quality control procedures. These procedures often involve an abundance cutoff for rare sequences or clustering of sequences, both of which limit genetic resolution. Here we propose a simple experimental protocol that retains the high genetic resolution granted by HTSeq methods while effectively removing many low abundance sequences that are likely due to PCR and sequencing errors. According to this protocol, we split samples and submit both halves to independent PCR and sequencing runs. The resulting sequence data is graphically and quantitatively characterized by the discordance between the two experimental branches, allowing for a quick identification of problematic samples. Further, we discard sequences that are not found in both branches ("AmpliconDuo filter"). We show that the majority of sequences removed in this way, mostly low abundance but also some higher abundance sequences, show features expected from random modifications of true sequences as introduced by PCR and sequencing errors. On the other hand, the filter retains many low abundance sequences observed in both branches and thus provides a more reliable census of the rare biosphere. We find that the AmpliconDuo filter increases biological resolution as it increases apparent community similarity between biologically similar communities, while it does not affect apparent community similarities between biologically dissimilar communities. The filter does not distort overall apparent community compositions. Finally, we quantitatively explain the effect of the AmpliconDuo filter by a simple mathematical model.
对小核糖体亚基扩增子进行高通量测序(HTSeq),有潜力全面表征微生物群落组成,甚至可以鉴定到稀有物种。然而,多步骤实验过程容易出错,这就要求对所得的原始序列进行质量控制程序。这些程序通常涉及对稀有序列设置丰度阈值或对序列进行聚类,这两者都会限制遗传分辨率。在此,我们提出一种简单的实验方案,该方案既能保留HTSeq方法所赋予的高遗传分辨率,又能有效去除许多可能由PCR和测序错误导致的低丰度序列。根据该方案,我们将样本分成两半,并将这两半分别进行独立的PCR和测序。通过两个实验分支之间的不一致性,以图形和定量方式对所得序列数据进行表征,从而快速识别有问题的样本。此外,我们舍弃在两个分支中都未出现的序列(“扩增子二元过滤器”)。我们表明,以这种方式去除的大多数序列,大多是低丰度序列,但也有一些高丰度序列,呈现出由PCR和测序错误对真实序列进行随机修饰所预期的特征。另一方面,该过滤器保留了在两个分支中都观察到的许多低丰度序列,因此能更可靠地普查稀有生物圈。我们发现,扩增子二元过滤器提高了生物学分辨率,因为它增加了生物学相似群落之间的表观群落相似性,而不影响生物学不相似群落之间的表观群落相似性。该过滤器不会扭曲整体表观群落组成。最后,我们通过一个简单的数学模型定量解释了扩增子二元过滤器的作用。