Dalm Virgil A S H, Driessen Gertjan J A, Barendregt Barbara H, van Hagen Petrus M, van der Burg Mirjam
Department of Internal Medicine, Erasmus MC, 's-Gravendijkwal 230, 3015 CE, Rotterdam, The Netherlands.
Department of Immunology, Erasmus MC, 's-Gravendijkwal 230, 3015 CE, Rotterdam, The Netherlands.
J Clin Immunol. 2015 Nov;35(8):761-8. doi: 10.1007/s10875-015-0211-z. Epub 2015 Nov 14.
Jacobsen syndrome (JS) is a rare contiguous gene syndrome caused by partial deletion of the long arm of chromosome 11. Clinical features include physical and mental growth retardation, facial dysmorphism, thrombocytopenia, impaired platelet function and pancytopenia. In case reports, recurrent infections and impaired immune cell function compatible with immunodeficiency were described. However, Jacobsen syndrome has not been recognized as an established syndromic primary immunodeficiency.
To evaluate the presence of immunodeficiency in a series of 6 patients with JS.
Medical history of 6 patients with JS was evaluated for recurrent infections. IgG, IgA, IgM and specific antibodies against S. pneumoniae were measured. Response to immunization with a polysaccharide vaccine (Pneumovax) was measured and B and T lymphocyte subset analyses were performed using flowcytometry.
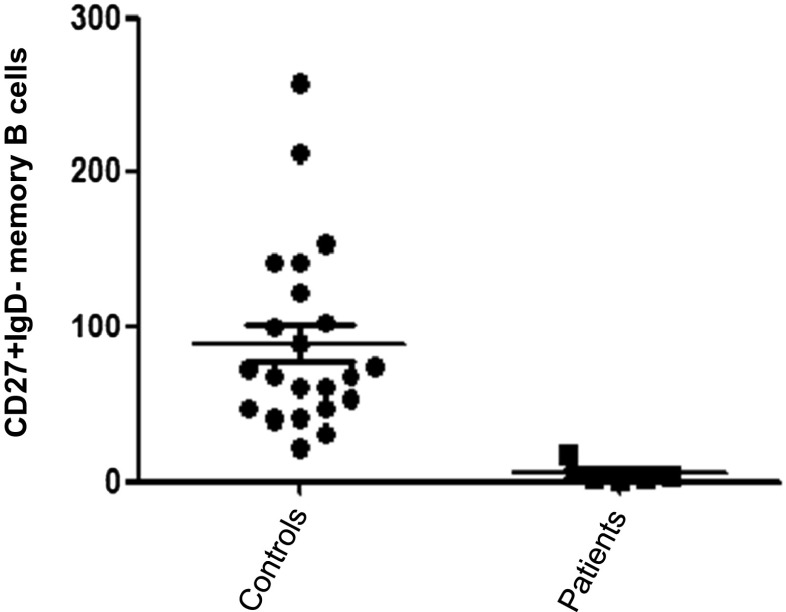
Five out of 6 patients suffered from recurrent infections. These patients had low IgG levels and impaired response to S. pneumoniae polysaccharide vaccination. Moreover, we also found a significant decrease in the absolute number of memory B cells, suggesting a defective germinal center function. In a number of patients, low numbers of T lymphocytes and NK cells were found.
Most patients with JS suffer from combined immunodeficiency in the presence of recurrent infections. Therefore, we consider JS a syndromic primary immunodeficiency. Early detection of immunodeficiency may reduce the frequency and severity of infections. All JS patients should therefore undergo immunological evaluation. Future studies in a larger cohort of patients will more precisely define the pathophysiology of the immunodeficiency in JS.
雅各布森综合征(JS)是一种由11号染色体长臂部分缺失引起的罕见的连续性基因综合征。临床特征包括身心发育迟缓、面部畸形、血小板减少、血小板功能受损和全血细胞减少。在病例报告中,描述了与免疫缺陷相符的反复感染和免疫细胞功能受损。然而,雅各布森综合征尚未被确认为一种既定的综合征性原发性免疫缺陷。
评估6例JS患者中免疫缺陷的存在情况。
评估6例JS患者的病史以了解反复感染情况。检测了IgG、IgA、IgM以及针对肺炎链球菌的特异性抗体。测定了对多糖疫苗(肺炎球菌疫苗)免疫接种的反应,并使用流式细胞术进行了B和T淋巴细胞亚群分析。
6例患者中有5例患有反复感染。这些患者的IgG水平较低,对肺炎链球菌多糖疫苗的反应受损。此外,我们还发现记忆B细胞的绝对数量显著减少,提示生发中心功能存在缺陷。在一些患者中,发现T淋巴细胞和NK细胞数量较少。
大多数JS患者在存在反复感染的情况下患有联合免疫缺陷。因此,我们认为JS是一种综合征性原发性免疫缺陷。早期发现免疫缺陷可能会降低感染的频率和严重程度。因此,所有JS患者都应接受免疫学评估。未来对更大患者队列的研究将更精确地界定JS中免疫缺陷的病理生理学。